Downloaded 84 times



![Connecting to Linux Startup the your machine and log in OR Remote connection (e.g. on departmental server) $ ssh [email_address] -> The sysadmin should have made you an account -> you are prompted for your password On windows, install PuTTY to connect http://www.chiark.greenend.org.uk/~sgtatham/putty/](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-4-2048.jpg)


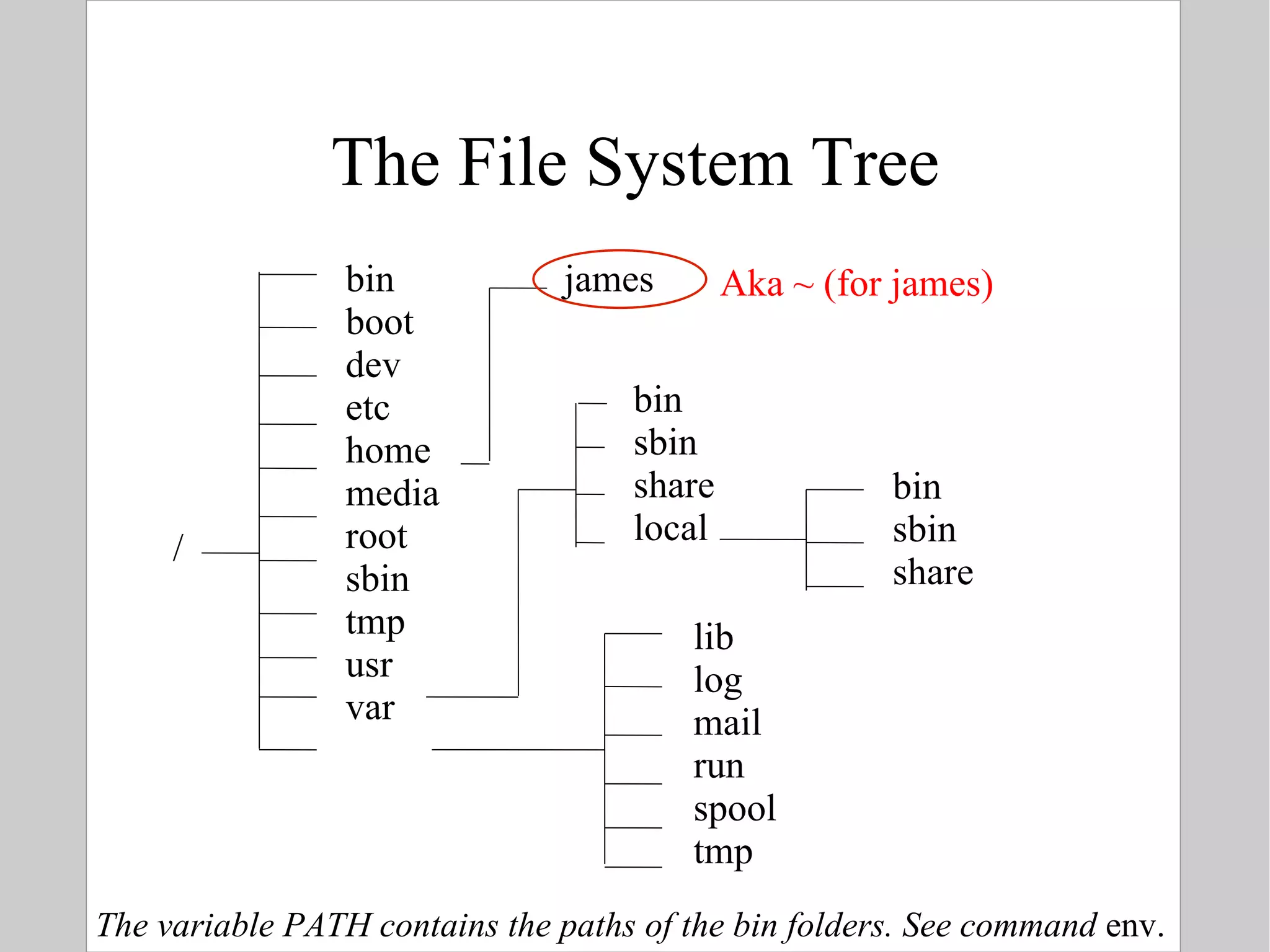

![Names of files & directories In UNIX names of files and directories are case sensitive Some characters have a special meaning to the shell: spaces, |, <, >, *, ?, [, ], /, \,.,.. You can use some of them, but they have to be hidden (escaped) from the shell In UNIX there is no such thing as file extensions : commands are marked executable via permissions files are recognized based on content Files and directories share the same name space](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-9-2048.jpg)


















![Remotely copying files If you are logged in on another machine, e.g. $ ssh [email_address] And you are working there: creating folders, files, running programs, you can copy to your own machine using scp $ scp [email_address] :/home/bits/sample.sam . Command to copy username machine Path to the remote folder / file you want to copy To the local current dir](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-28-2048.jpg)

![Solutions $ scp [email_address] :/home/bits/TAIR9_mRNA.bed . $ scp [email_address] :/home/bits/recut . $ apropos download $ wget http://dl.dropbox.com/u/18352887/BITS_training_material/Link%20to%20sample.sam $ mkdir -p bioinfo/{data,bin} $ mv *.sam bioinfo/data $ mv *.bed bioinfo/data $ mv recut bioinfo/bin](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-30-2048.jpg)















![Compression To compress one or more files: $ gzip [ options ] file $ bzip2 [ options ] file Options: c: send output to stdout instead of overwriting the specified file(s) 1 or --fast: fast / minimal compression 9 or --best: slow / maximal compression Standard extensions: gzip .gz bzip2 .bz2](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-51-2048.jpg)
![Decompression To decompress one or more files: $ gunzip [ options ] file(s) $ bunzip2 [ options ] file(s) To decompress a tar.gz or tar.bz2 $ tar xvfz file.tar.gz $ tar xvfj file.tar.bz2 The following tools can read directly from gzip or bzip2 files (*.bz2 or *.gz) $ zcat file(s) $ bzcat file(s)](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-52-2048.jpg)



![grep grep is used to extract lines from an input stream that match (or don't match) a regular expression Syntax: $ grep [ options ] regex [ file(s) ] The file(s) (or if omitted stdin ) are read line by line. If the line matches the given criteria, the entire line is written to stdout.](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-56-2048.jpg)




![Word Count A general tool for counting lines, words and characters: wc [ options ] file(s) Interesting options: c : number of characters w : number of words l : number of lines Example: How many packages are installed? $ yum list installed | wc -l](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-61-2048.jpg)
![Transform To manipulate individual characters in an input stream: $ tr 's1' 's2' ! tr always reads from stdin – you cannot specify any files as command line arguments Characters in s1 are replaced by characters in s2 The result is written to stdout Example: $ echo 'James Watson' | \ tr '[a-z]' '[A-Z]' JAMES WATSON](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-62-2048.jpg)






![cut A similar tool is cut , it extracts fields from fixed text file formats only: fixed width $ cut -c LIST [ file ] fixed delimiter $ cut [-d delim ] -f LIST [ file ] For LIST: N : the Nth element N-M : element the Nth till the Mth element N- : from the Nth element on -M : till the Mth element The first element is 1](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-69-2048.jpg)


![sort To sort alphabetically or numerically lines of text: $ sort [ options ] file(s) When one or more file(s) are specified, they are read one by one, but all lines are sorted. The output is written to stdout When no file(s) arguments are given, sort reads input from stdin](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-72-2048.jpg)





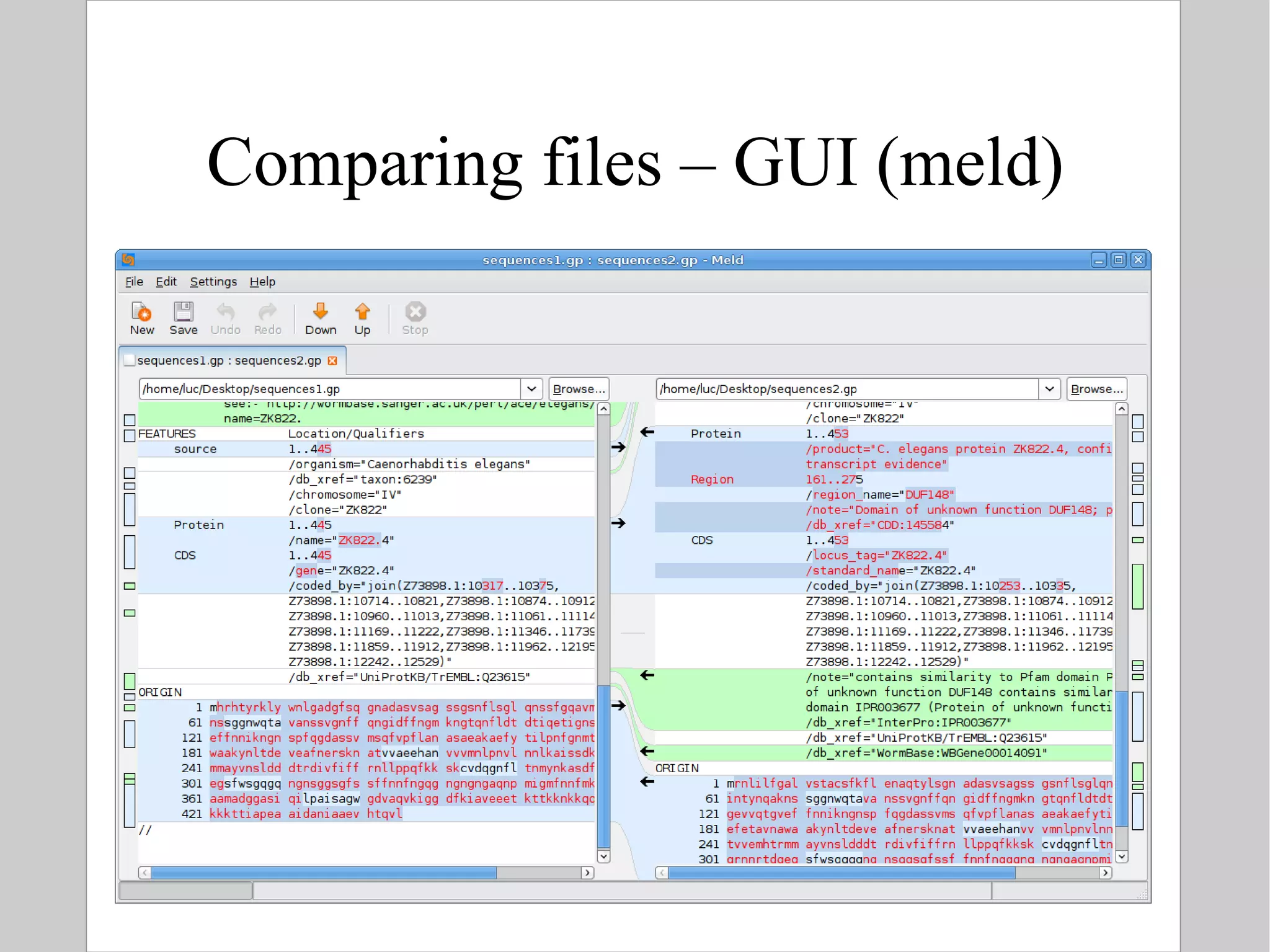
![Comparing text files To find differences between two text files: $ diff [ options ] file1 file2 Example: difference between two genbank versions of LOCUS CAA98068 # diff sequences1.gp sequences2.gp 1,2c1,3 < LOCUS CAA98068 445 aa linear INV 27-OCT-2000 < DEFINITION ZK822.4 [Caenorhabditis elegans]. --- > LOCUS CAA98068 453 aa linear INV 09-MAY-2010 > DEFINITION C. elegans protein ZK822.4, confirmed by transcript evidence > [Caenorhabditis elegans]. 4,5c5,6 < VERSION CAA98068.1 GI:3881817 < DBSOURCE embl locus CEZK822, accession Z73898.1 --- > VERSION CAA98068.2 GI:14530708 > DBSOURCE embl accession Z73898.1](https://image.slidesharecdn.com/introlinux3textminingtools-110628052526-phpapp01/75/BITS-Introduction-to-Linux-Text-manipulation-tools-for-bioinformatics-78-2048.jpg)







The document provides an introduction to using the Linux command line for bioinformatics tasks. It covers navigating the file system, manipulating files and directories, input/output redirection, piping commands together, and commonly used text processing tools. The goal is to help users easily use command line tools, automate repetitive tasks, and parse/summarize text-based outputs.























































































