

miR-34a(-/-) mice are susceptible to diet-induced obesity
- PMID: 27377585
- PMCID: PMC4979678
- DOI: 10.1002/oby.21561
miR-34a(-/-) mice are susceptible to diet-induced obesity
Abstract
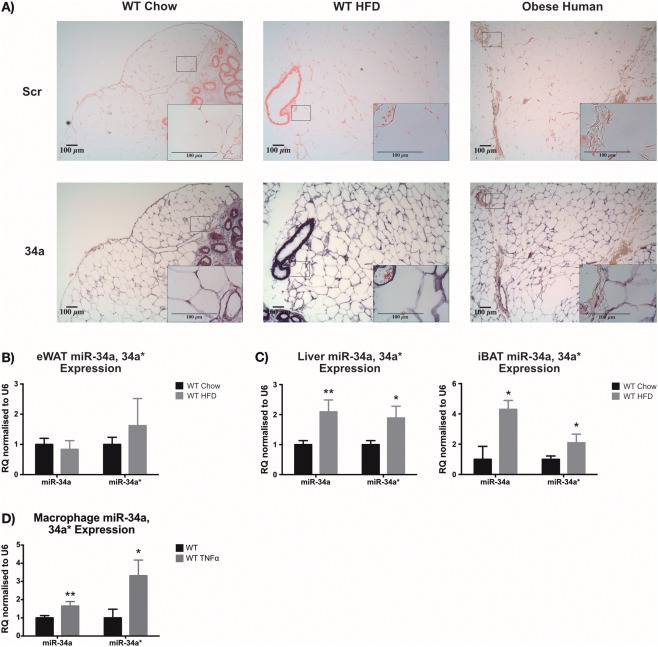
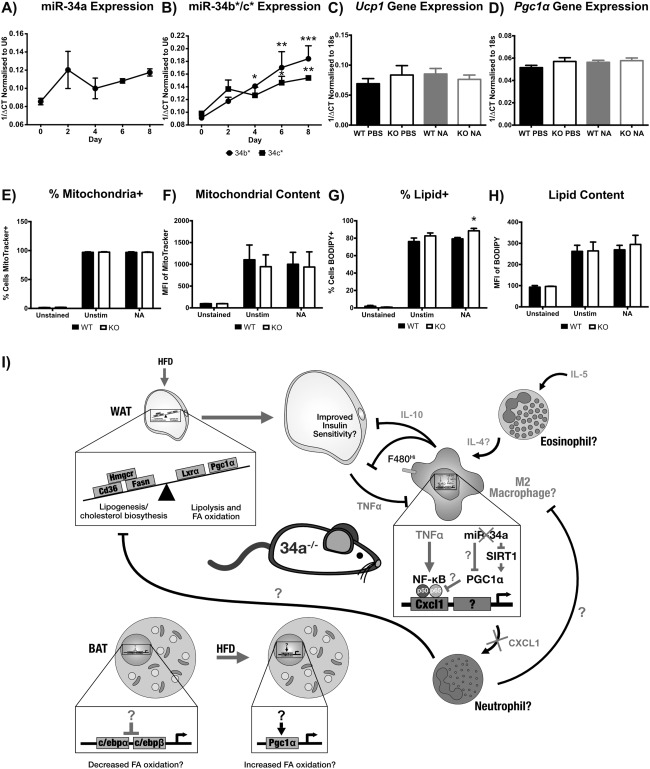
Objective: MicroRNA (miR)-34a regulates inflammatory pathways, and increased transcripts have been observed in serum and subcutaneous adipose of subjects who have obesity and type 2 diabetes. Therefore, the role of miR-34a in adipose tissue inflammation and lipid metabolism in murine diet-induced obesity was investigated.
Methods: Wild-type (WT) and miR-34a(-/-) mice were fed chow or high-fat diet (HFD) for 24 weeks. WT and miR-34a(-/-) bone marrow-derived macrophages were cultured in vitro with macrophage colony-stimulating factor (M-CSF). Brown and white preadipocytes were cultured from the stromal vascular fraction (SVF) of intrascapular brown and epididymal white adipose tissue (eWAT), with rosiglitazone.
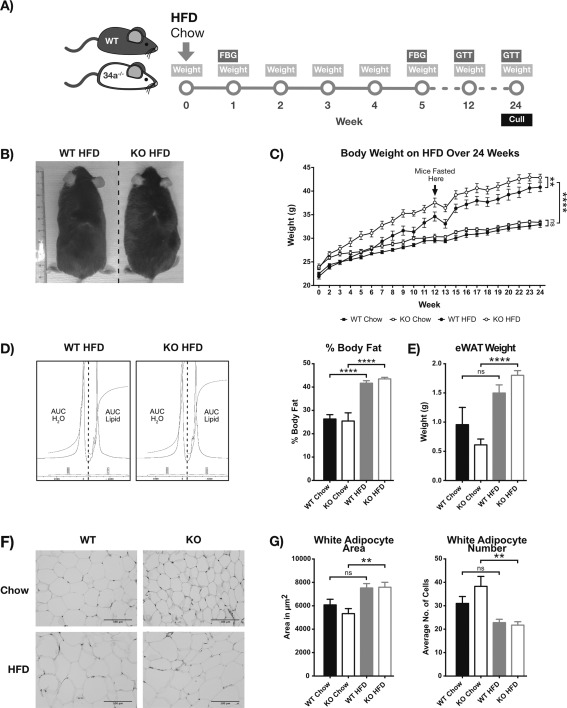
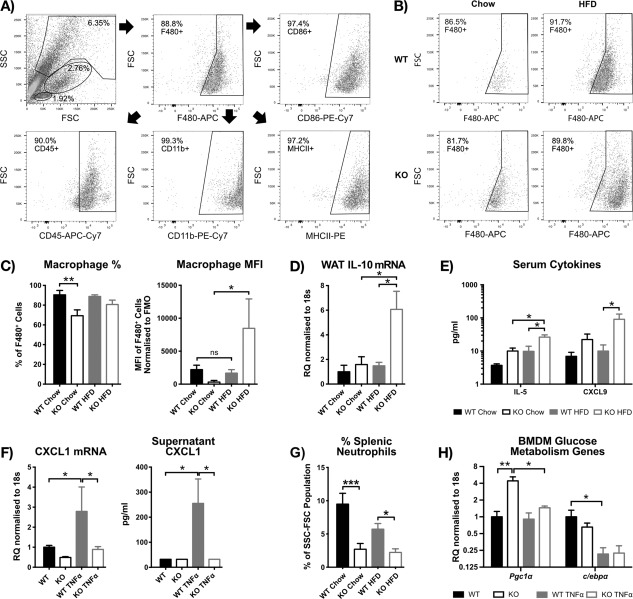
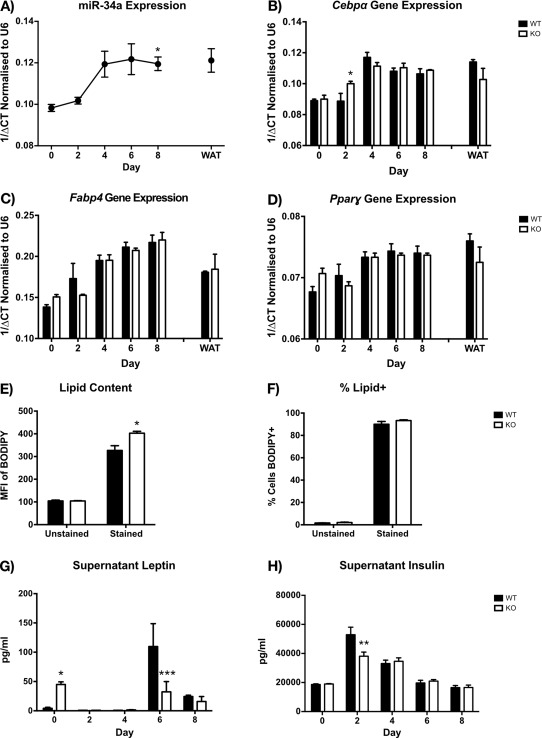
Results: HFD-fed miR-34a(-/-) mice were significantly heavier with a greater increase in eWAT weight than WT. miR-34a(-/-) eWAT had a smaller adipocyte area, which significantly increased with HFD. miR-34a(-/-) eWAT showed basal increases in Cd36, Hmgcr, Lxrα, Pgc1α, and Fasn. miR-34a(-/-) intrascapular brown adipose tissue had basal reductions in c/ebpα and c/ebpβ, with in vitro miR-34a(-/-) white adipocytes showing increased lipid content. An F4/80(high) macrophage population was present in HFD miR-34a(-/-) eWAT, with increased IL-10 transcripts and serum IL-5 protein. Finally, miR-34a(-/-) bone marrow-derived macrophages showed an ablated CXCL1 response to tumor necrosis factor-α.
Conclusions: These findings suggest a multifactorial role of miR-34a in controlling susceptibility to obesity, by regulating inflammatory and metabolic pathways.
© 2016 The Obesity Society.
Figures

References
-
- Gregor M. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011;29:415. - PubMed
-
- Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 2005;96:939–949. - PubMed
-
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev 2004;84:277–359. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
