

Autophagy in neuroprotection and neurodegeneration: A question of balance
- PMID: 18806889
- PMCID: PMC2544613
- DOI: 10.2217/14796708.3.3.309
Autophagy in neuroprotection and neurodegeneration: A question of balance
Abstract
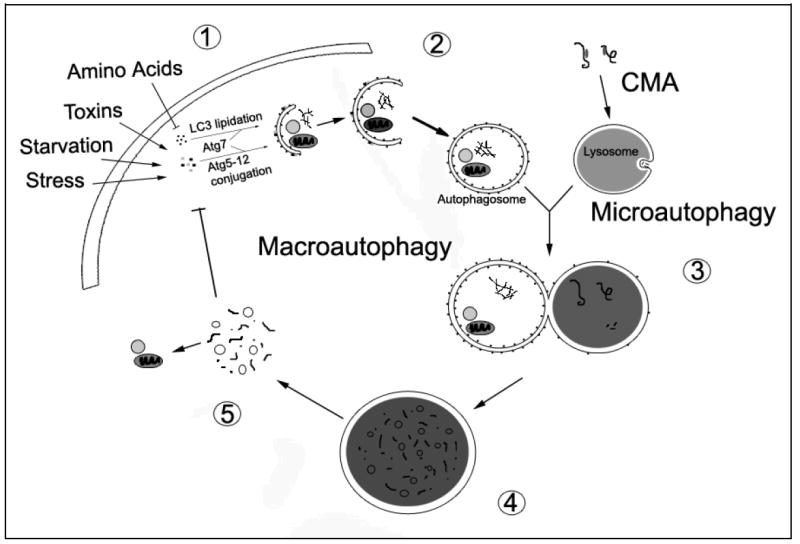
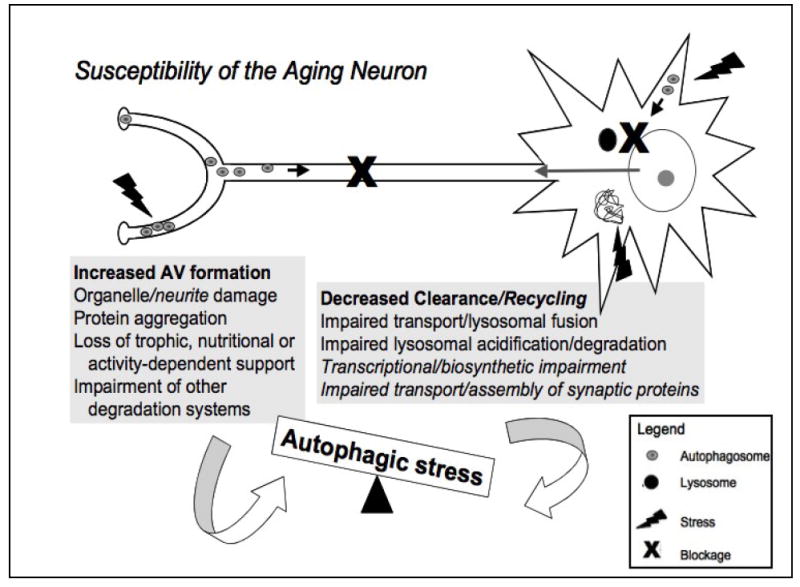
A central issue in developing therapies for neurodegenerative diseases involves understanding why adaptive responses to stress or injury fail to prevent synaptic dysfunction and neuronal cell death. Macroautophagy is a major, evolutionarily conserved response to nutrient and bioenergetic stresses, which has the capacity to remove aggregated proteins and damaged organelles such as mitochondria. This has prompted intense interest in autophagy-related therapies for Huntington's, Alzheimer's, Parkinson's, stroke and other neurological diseases. However, excessive or imbalanced induction of autophagic recycling can actively contribute to neuronal atrophy, neurite degeneration and cell death. Oxidative-, aging- and disease-related increase in demand for autophagy, coupled with declining axonal trafficking, lysosomal degradation or biosynthetic efficiencies promote increased susceptibility to a harmful state of autophagic stress. A more complete understanding of dysfunction along the entire spectrum of autophagic recycling, from autophagosome formation through clearance and regeneration of new cellular components is necessary to restore balance to the system, promote neuronal health and maximize therapeutic potentials.
Figures

References
-
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. - PubMed
-
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. - PubMed
-
- Bandhyopadhyay U, Cuervo AM. Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp Gerontol. 2007;42:120–8. - PubMed
-
- Farre JC, Subramani S. Peroxisome turnover by micropexophagy: an autophagy-related process. Trends Cell Biol. 2004;14:515–23. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources