

Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells
- PMID: 12511591
- PMCID: PMC151834
- DOI: 10.1172/JCI16147
Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells
Abstract
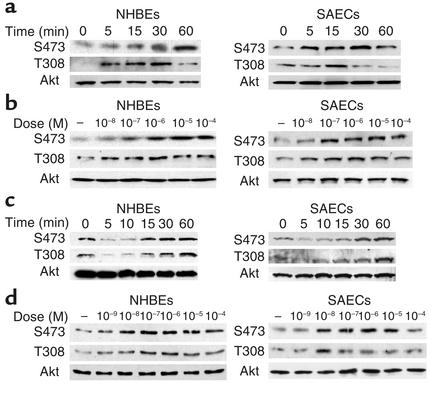
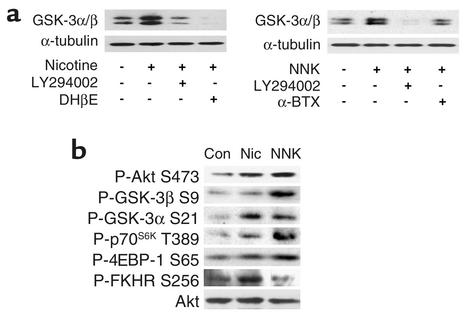
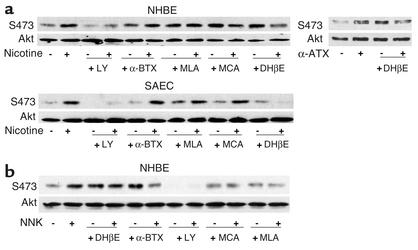
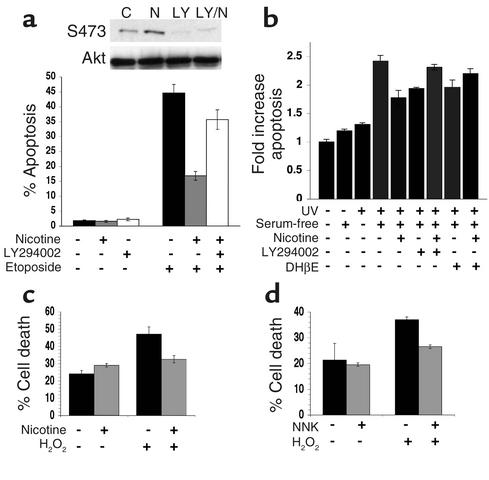
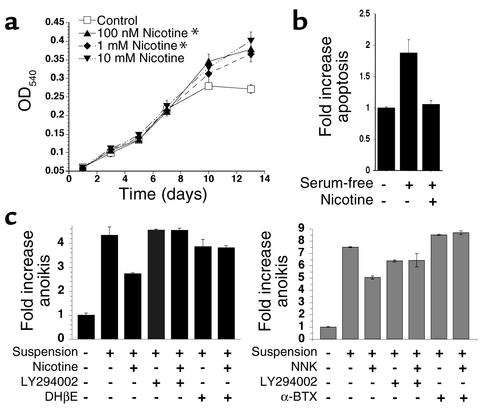
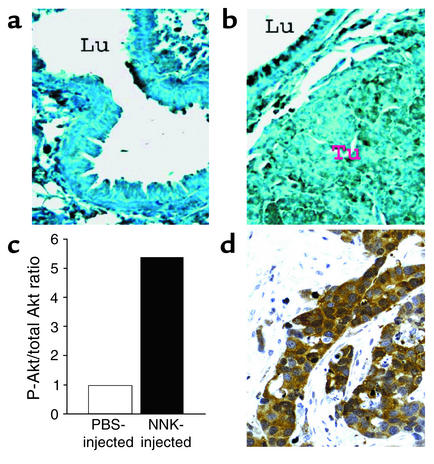
Tobacco-related diseases such as lung cancer cause over 4.2 million deaths annually, with approximately 400,000 deaths per year occurring in the US. Genotoxic effects of tobacco components have been described, but effects on signaling pathways in normal cells have not been described. Here, we show activation of the serine/threonine kinase Akt in nonimmortalized human airway epithelial cells in vitro by two components of cigarette smoke, nicotine and the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Activation of Akt by nicotine or NNK occurred within minutes at concentrations achievable by smokers and depended upon alpha(3)-/alpha(4)-containing or alpha(7)-containing nicotinic acetylcholine receptors, respectively. Activated Akt increased phosphorylation of downstream substrates such as GSK-3, p70(S6K), 4EBP-1, and FKHR. Treatment with nicotine or NNK attenuated apoptosis caused by etoposide, ultraviolet irradiation, or hydrogen peroxide and partially induced a transformed phenotype manifest as loss of contact inhibition and loss of dependence on exogenous growth factors or adherence to ECM. In vivo, active Akt was detected in airway epithelial cells and lung tumors from NNK-treated A/J mice, and in human lung cancers derived from smokers. Redundant Akt activation by nicotine and NNK could contribute to tobacco-related carcinogenesis by regulating two processes critical for tumorigenesis, cell growth and apoptosis.
Figures
Comment in
-
Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer.J Clin Invest. 2003 Jan;111(1):31-3. doi: 10.1172/JCI17492. J Clin Invest. 2003. PMID: 12511585 Free PMC article. Review. No abstract available.
References
-
- Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001;114:2903–2910. - PubMed
-
- Scheid MP, Woodgett JR. Pkb/akt: functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001;2:760–768. - PubMed
-
- Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61:3986–3997. - PubMed
-
- Maus AD, et al. Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol. Pharmacol. 1998;54:779–788. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
