

Abstract
There is an expanding need to modify plant genomes to create new plant germplasm that advances both basic and applied plant research. Most current methods for plant genome modification involve regenerating plants from genetically modified cells in tissue culture, which is technically challenging, expensive and time consuming, and works with limited plant species or genotypes. Herein, we describe two Agrobacterium-based methods for creating genetic modifications on either sterilely grown or soil-grown Nicotiana benthamiana plants. These methods use developmental regulators (DRs), gene products that influence cell division and differentiation, to induce de novo meristems. Genome editing reagents, such as the RNA-guided endonuclease Cas9, may be co-delivered with the DRs to create shoots that transmit edits to the next generation. One method, called fast-treated Agrobacterium co-culture (Fast-TrACC), delivers DRs to seedlings grown aseptically; meristems that produce shoots and ultimately whole plants are induced. The other approach, called direct delivery (DD), involves delivering DRs to soil-grown plants from which existing meristems have been removed; the DRs promote the formation of new shoots at the wound site. With either approach, if transgene cassettes and/or gene editing reagents are provided, these induced, de novo meristems may be transgenic, edited or both. These two methods offer alternative approaches for generating novel plant germplasm that are cheaper and less technically challenging and take less time than standard approaches. The whole procedure from transfer DNA (T-DNA) assembly to recovery of edited plants can be completed in ~70 d for both DD and Fast-TrACC.
Introduction
In plants, conventional genetic engineering methods most commonly use Agrobacterium tumefaciens or biolistics to deliver transgenes to cells1. The resulting transgenic cells are subsequently selected and regenerated into whole plants using tissue culture techniques. For the past 30 years, this transformation process has facilitated plant improvement by expanding available traits, leading to increased agricultural productivity, and fueled the discovery of new biological mechanisms. The advent of gene editing technologies, such as clustered regularly interspaced short palindromic repeats-Cas9, provides additional approaches to modify genomes, including inactivating or replacing genes. However, to implement gene editing, researchers are still typically required to use traditional plant transformation protocols to introduce foreign DNA into plant cells; these methods can take 6–12 months in many crop species. Faced with future challenges of creating a sustainable food supply and adapting crops to dynamic environmental conditions, there is a need for innovations that expedite the production of plants genetically modified through transgenesis and gene editing.
Previous work has shown that ectopic expression of regulatory genes that control specific developmental pathways can accelerate the production of transgenic and gene-edited plants through tissue culture2–4. For example, delivery of maize Wuschel2 (Wus2) and Baby Boom (Bbm)—transcription factors involved in meristem maintenance and identity—promotes the production of somatic embryos and increases transformation efficiencies in previously nontransformable inbred maize lines5. However, constitutive expression of Wus2 and Bbm results in phenotypic abnormalities upon regeneration of transgenic plants, requiring removal with site-specific recombinases or controlled expression using tissue-specific promoters2,5. In addition to Wus2 and Bbm, a number of different regulatory genes of both plant and nonplant origin have been applied in a variety of species for the purpose of enhancing plant transformation4. We collectively refer to these genes as developmental regulators (DRs).
Building on this work, our laboratory developed two methods that involve inducing meristems on dicotyledonous plants6. Meristems are created by delivering DRs such as Wus2, Bbm or genes involved in the synthesis of cytokinin. One method, called fast-treated Agrobacterium co-culture (Fast-TrACC), delivers DRs to seedlings grown aseptically; meristems that produce shoots and ultimately whole plants are induced. The other approach, called direct delivery (DD), involves delivering DRs to soil-grown plants from which existing meristems have been removed; the DRs promote the formation of new shoots at the wound site. With either approach, if transgene cassettes and/or gene-editing reagents are provided, these induced, de novo meristems may be transgenic, edited or both6. The genetically modified meristems may also transmit modifications to the next generation.
In the context of plant transformation and gene editing, the DD and Fast-TrACC methods provide novel alternatives to traditional methodologies. Here, we describe in detail how to carry out these methods in Nicotiana benthamiana, the first dicotyledonous species for which these methods have been optimized. We assume that additional optimization of delivery methods and expression of the DRs will be required if researchers attempt to adopt the protocols for other plant species.
Procedure overview
Cloning methods described herein cover the production of Agrobacterium transformation vectors with expression cassettes for single guide RNAs (sgRNAs) and DRs for use in both the DD and Fast-TrACC methods (Fig. 1). Steps 1–18 of Procedure 1 describe the use of a modular cloning (MoClo) strategy for assembling sgRNA arrays for the expression of multiple guides from a single RNA polymerase II promoter (Fig. 2a,b and Supplementary Tables 1 and 2). These sgRNA cassettes are flanked by transfer RNA (tRNA) sequences that are processed by endogenous enzymes to release individual sgRNAs7. Steps 19–21 of Procedure 1 combine these sgRNA-tRNA expression vectors with available DR expression cassettes (Supplementary Table 3) to assemble final T-DNA constructs (Fig. 2c). These cloning protocols are built on systems previously described in refs. 8,9. Assembly of the final T-DNA vector uses existing modules and backbone vectors or custom modules that express any gene of interest (Supplementary Tables 3 and 4). New DNA modules are compatible with the three-component A/B/C cloning system previously described by Cermak et al.8, and additional vectors create a four-component A/B/C′/D cloning system (Figs. 1 and 2c). Additionally, backbone vectors contain bean yellow dwarf virus (BeYDV) replicon components previously described by Baltes et al.10. Steps 1–21 of Procedure 2 describe the use of DR transformation vectors in the DD method, whereas Steps 1–57 of Procedure 3 describe their use in Fast-TrACC.
Fig. 1 |. Overview of procedures.

a, Procedure 1, Golden Gate assembly of the T-DNA vector. Four modules (A–D) encode genes of interest, such as reporters (A), gene-editing reagents (A and/or B) and DRs (C′ and D). The modules are cloned into T-DNA vectors using Golden Gate assembly and used in downstream DD or Fast-TrACC procedures. b, Procedure 2, DD: (1) All meristems are removed from mature N. benthamiana plants grown in soil. (2) Agrobacterium cultures are delivered to trimmed plants. (3) De novo meristems are induced, and new shoots contain genetic modifications of interest. c, Procedure 3, Fast-TrACC: (1) N. benthamiana seeds are sown into each well of a six-well plate. (2) Five days after germination, the seedlings are co-cultured with Agrobacterium strains. (3) The seedlings are rinsed, and individual seedlings are transferred to wells in a 12-well plate. Growths form on the cotyledons due to DR overexpression; some of these growths produce shoots. (4) Shoot-like growths are excised from parent seedlings and transferred to solid medium. (5) Shoots from the solid medium are transferred to rooting medium. (6) Fully regenerated and genetically modified plants are transferred to soil.
Fig. 2 |. Golden Gate assembly of T-DNA vectors.
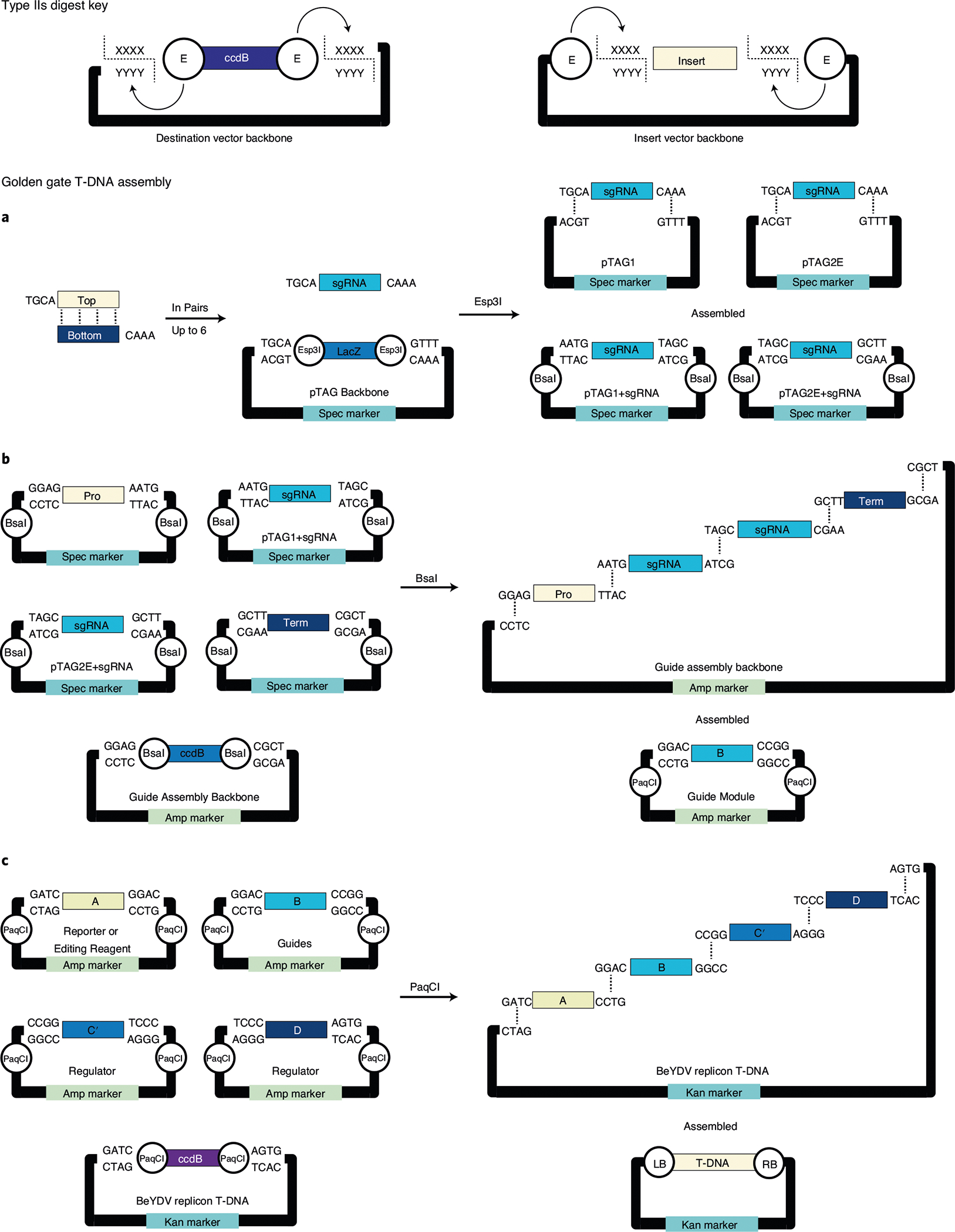
Golden Gate modular assembly takes advantage of type IIs restriction enzymes, which cleave DNA outside recognition sites. The type IIs digest key describes the directionality of enzyme cutting with respect to position on cloning vectors used in Procedure 1. Enzymes (E) digest destination vectors away from CcdB and toward inserts, which prime vectors for assembly in Golden Gate reactions. a–c, Golden Gate cycles from Procedure 1, where assembly of inserts into destination vectors changes antibiotic selectable markers (spectinomycin, ampicillin and kanamycin) and type IIs enzymes used in subsequent reactions. Changing selectable markers and type IIs enzymes is important in reducing background and changing overhangs used in downstream assemblies, respectively. Each panel outlines all overhangs in each Golden Gate reaction and assembled vectors with new type IIs enzymes used in downstream assemblies. Positional cloning of two sgRNAs (a). Even numbered (two, four or six) sets of sgRNAs are individually assembled into pTAG vector backbones. Complementary oligos for sgRNA spacer sequences are annealed, leaving compatible overhangs for vector entry. The sgRNA vector backbones contain tRNA processing sequences that release individual sgRNAs when expressed in the cell (TAG vectors: pTAG1-pTAG6E). The sgRNAs are assembled into the backbone using the type IIs restriction enzyme, Esp3I. Assembly of the sgRNA-tRNA array in module B (b). Previously assembled sgRNA vectors (a) are positionally cloned between a RNA Pol II promoter (Pro) and terminator (Term) of choice (Supplementary Table 2) using the type IIs restriction enzyme, BsaI. Individual sgRNAs in the array are separated by tRNA processing sequences contributed by the backbones in the previous cloning step. The expression cassette is assembled into a B module vector (typically) for downstream modular cloning. Assembly of the transformation vector (c). Inserts from modules A, B, C′ and D are sequentially assembled into a final T-DNA backbone using the type IIs enzyme, PaqCI. Activity of PaqCI removes the bacterial negative selectable marker, CcdB, from the transformation vector backbone. The T-DNA contains BeYDV replicon components, previously described in ref. 10, between the left (LB) and right (RB) T-DNA borders.
Given that optimizing Fast-TrACC and DD in different plant species and implementing strategies for DR removal will require researchers to create and test a variety of different constructs, we include a protocol for high-throughput, modular assembly of T-DNAs using Golden Gate cloning (Procedure 1). Supplementary Table 5 lists a collection of preassembled T-DNA vectors that can be used to quickly test the effectiveness of both the DD and Fast-TrACC methods using existing reagents in various plant species. If different reporters are desired, researchers can select specific A, B, C′ and D modules in Supplementary Tables 3 and 4 and follow Steps 19–21 in Procedure 1 to create new T-DNA vectors in ~1 week.
Applications of methods
Fast-TrACC is a highly scalable method that is quick to implement and requires little plant growth space. This makes it possible to rapidly test variables to optimize gene delivery and subsequent meristem induction in different plant species. These variables include, but are not limited to, Agrobacterium strains for DR delivery, combinations of DRs to best establish regenerative competency, modifications to DR coding sequences that enhance function and the use of different promoters to tune DR expression. Whereas the complete regeneration of an edited plant via Fast-TrACC takes ~70 d, the assessment of different variables for meristem induction takes only ~20 d after the assembly and transformation of test vectors into Agrobacterium.
DD is performed on whole plants in nonsterile conditions, circumventing the aseptic culture steps required for most conventional plant transformation protocols. This aspect of DD makes it attractive for use on plant species that are less responsive to growth in culture; however, its deployment in other species will likely require additional protocol optimization. DD can be carried out without special equipment and reagents, such as a laminar flow hood and tissue culture media. For laboratories on a budget, DD is an inexpensive approach for producing genetically modified plants.
Maher et al. created double biallelic knockout mutations in phytoene desaturase homologs without transgene integration6. These experiments were performed using transgenic N. benthamiana lines that constitutively express Cas9. In unpublished work, we have used wild-type plants to create gene edits wherein both Cas9 and the sgRNAs are delivered by the T-DNA. Additional applications of Fast-TrACC and DD have yet to be explored. For example, multiplexed sgRNA arrays could be used to create mutations in multiple genomic targets. Additionally, the use of more sophisticated editing technologies, such as base editors or prime editors, could expand possible applications11. In the context of creating transgenics, co-delivery of ‘trait vectors’ together with DR T-DNAs could be used to recover stable insertions without phenotypic abnormalities4. Also, through the exploitation of homology-directed DNA repair mechanisms, Cas9-mediated gene targeting may be possible using both DD and Fast-TrACC, though this approach has not yet been demonstrated.
Alternative methods
Conventional methods of plant transformation generally use plant tissues cultured aseptically in vitro, and DNA is delivered to such tissue via particle bombardment or Agrobacterium. DNA delivery is typically followed by steps of callus induction, selection for transgenic cells and then regeneration steps to produce viable plants where stable insertions or edits are heritable. Many plant species are amenable to this process; however, the cell types used to derive callus are variable12 and include zygotic embryos13, cotyledons14,15 and true leaves16. Other methods of in vitro transformation involve polyethylene glycol or electroporation-mediated transfection of DNA into single-cell protoplasts followed by regeneration steps; however, protoplast regeneration is limited to a few plant species, such as lettuce17, tobacco18 and rice19.
Whereas conventional in vitro culture methods are sometimes robust, there are several limitations that reduce the transformation efficiency and narrow the scope of genotypes that can be used for gene editing or the production of stable, genetically modified plant lines1. Many agronomically important crops species, such as cereals, legumes, woody plants and certain elite cultivars, are recalcitrant to conventional in vitro culture techniques, owing to a lower response on hormone media for shoot and root regeneration12. To overcome this, researchers have taken alternative approaches for transformation, including the use of morphogenic regulator genes4, such as Wus2 and Bbm, or in planta methods that deliver transgenes to other tissues, such as the embryonic axis or the plumular meristem of seedlings20–22 or the axillary meristems of developed plants6. While these in planta methods use strategies similar to those described herein, such as seedling infection or the use of a needle and syringe, differences exist in the developmental stage of the targeted tissue or cells, the use of regulator genes and the experimental outcomes (i.e. stable insertion of T-DNAs or editing in the absence of editing reagents).
The protocols described herein provide robust alternatives for genetically modifying N. benthamiana compared with traditional, tissue culture-based transformation methods23. These protocols are particularly appropriate for laboratories that do not have in-house expertise on plant tissue culture or are interested in developing tissue culture-free methods of plant transformation in species that are recalcitrant to regeneration through tissue culture.
Limitations
Whereas the expression of DRs for meristem induction holds much promise for creating new plant germplasm, there are also inherent limitations that must be considered, particularly when applying these methods to species other than N. benthamiana. The ability of DRs to induce organogenesis in a given tissue depends on species-specific expression cascades that regulate plant patterning24. Applications in other species will likely require the optimization of a number of variables, including the DR reagent expression (both magnitude and spatiotemporal expression), Agrobacterium-mediated transformation (which is impacted by a species’ susceptibility and defense response to Agrobacterium) and growth/culture conditions, among others. The descriptions of reagents and their use in this protocol should be applied as general guidelines that can be optimized if applied to another plant species.
An important limitation to consider is that ectopic expression of DRs can have potentially deleterious impacts on plant growth, fertility and overall viability. These pleiotropic effects may impact regeneration efficiencies4–6. To minimize this issue, researchers working with the regulators Wus2 and Bbm to enhance maize transformation efficiencies have devised rab17, to drive expression of Cre recombinase, an enzyme that removes DR expression cassettes flanked by loxP sites5. While recombinase-mediated removal is effective, further enhancements were achieved by using the promoters PLTP and Axig1 for the expression of Wus2 and Bbm, respectively. These promoters not only reduced the regulator expression levels in developing tissues but also increased the rate of somatic embryo formation2. Additionally, Gordon-Kamm et al. demonstrated that the noncell autonomous nature of Wus2 alone was sufficient to stimulate somatic embryo formation in neighboring cells4. If transgenes of interest were co-delivered with T-DNAs carrying DR genes, the activity of Wus2 in trans could stimulate the formation of transgenic plants without the stable integration of DRs.
Experimental design
Golden Gate assembly of T-DNA vectors (Procedure 1, Steps 1–21)
Procedure 1 will enable researchers to quickly generate a collection of T-DNA vectors to test various methods of transformation and to deliver gene-editing reagents or reporters. Steps 1–14 of Procedure 1 describe the cloning of Cas9 sgRNA-tRNA arrays. This array cloning strategy allows the assembly of up to six different guide RNA sequences expressed from a single RNA polymerase II promoter in even-numbered (two, four or six) sets of sgRNAs. The successful use of pol II promoters to drive the expression of polycistronic sgRNA-tRNA transcripts has been demonstrated previously by the Voytas laboratory8. Alternatively, single sgRNA modules expressed by pol III promoters are described in Cermak et al. and all relevant vectors are distributed by Addgene. The methods described herein utilize an alternative, two-step cloning approach that is compatible with certain level 0 MoClo vectors9. This system enables the subsequent assembly of a final T-DNA vector using DR expression modules that are compatible with the Voytas laboratory’s previously published multipurpose genome engineering toolkit for plants8. Modules described as C′ must be used with a module D in a four-part assembly and are not interchangeable with previously described C modules.
Agrobacterium strain selection (for Procedure 1, Steps 22–27)
The DD and Fast-TrACC protocols describe the use of Agrobacterium strains AGL1, EHA105 and GV3101. A given strain may be more efficient for transformation depending on the plant species. It is recommended that researchers review the literature to determine which Agrobacterium strain should be used for a given species. For N. benthamiana, GV3101 works well in both the DD and Fast-TrACC protocols. For selection on solid medium, Agrobacterium strains AGL1 and EHA105 require 50 mg l−1 kanamycin and 10 mg l−1 rifampicin for selection. GV3101 requires 50 mg l−1 kanamycin, 50 mg l−1 gentamycin and 10 mg l−1 rifampicin.
DR infection strategy
For DD, isopentenyl transferase (ipt) alone is necessary and sufficient for inducing shoots on N. benthamiana. Combinations with Wus2 (whether using homologs from Zea mays or other species) have not been confirmed to enhance shoot formation relative to ipt alone. For Fast-TrACC, Wus2 is necessary and sufficient to induce growths on N. benthamiana, but shoot induction heavily depends on the expression of ipt.
We have explored using a ‘split vector’ transformation strategy, wherein multiple T-DNAs expressing DRs and/or gene-editing reagents are co-delivered to N. benthamiana at 1:1 or 1:1:1 ratio. Since constitutive expression of DRs can lead to negative pleiotropic effects on plant growth and development, this strategy makes it possible to recover transformed or gene-edited plants without the integration of DR expression constructs25. To apply this strategy in Procedures 2 and 3, multiple T-DNA vectors must be generated in Procedure 1. To accomplish this, we provide ‘empty 0000’ modules that can be used in the final T-DNA assembly (Fig. 2c and Supplementary Table 3). For example, if researchers want to generate a T-DNA vector that only contains Wus2 and the assembled sgRNA array (Steps 1–18, Procedure 1), this could be accomplished by selecting pMOD_A0000, pMOD_C’5014, pMOD_D0000 and pTrans_201 from Supplementary Table 3 for use in Steps 19–20, Procedure 1.
Choice of DD protocol (Procedure 2)
DD can be used in scenarios where researchers desire transgene delivery and subsequent editing without growing plants aseptically. Agrobacterium delivery to sites of pruning and subsequent expression of DRs stimulate the formation of de novo meristems.
Choice of Fast-TrACC protocol (Procedure 3)
As described by Maher et al.6, Fast-TrACC can be used to create both transgenic and gene-edited lines. Fast-TrACC can also be used to achieve transient gene expression, for example, to assess promoter activity or sgRNA function in infected seedlings after co-cultivation (Procedure 3, Step 33).
Controls (Procedures 2 and 3)
DD and Fast-TrACC experiments should include negative (no infection) controls. For DD, researchers should inject ~3 plants with injection medium only, or ideally with an Agrobacterium strain that harbors no T-DNA. These negative controls make it possible to measure the degree of background shooting that occurs during plant maintenance and the de novo shoot formation process (Steps 17 and 18, Procedure 2). In Fast-TrACC, ~10–15 seedlings should be treated with only co-cultivation medium or infected with an Agrobacterium strain lacking T-DNA. These seedlings can be compared with experimental treatments during the growth development phase (Steps 34–38, Procedure 3).
Protocol adaptation for alternative species
For researchers exploring the possibility of using in planta transformation methods to circumvent tissue culture, it is important to review existing literature before investing time in experiments. It is possible that certain techniques will work better than the methods presented in this manuscript (see Alternative methods section for further discussion). Maher et al.6 present proof-of-concept transformation methods that have only been demonstrated to work in N. benthamiana. The adaptation of such protocols to other species may require changes to the experimental procedure. These could include, but are not limited to, the use of regulator genes, the strategy of Agrobacterium infection (i.e., the cell type or the developmental stage of the plant), the location of callus-like growth formations and the timing of the transformation protocol. In the field of plant transformation, there are no standard procedures that work across all species. The purpose of this manuscript is to serve as a starting point to establish protocols for plants that are more difficult to transform using conventional in vitro methods.
Materials
▲CRITICAL To reduce exposure to potential chemical hazards, follow general laboratory practices and refer to the MSDS sheet for each chemical before use ▲CRITICAL Unless otherwise noted, reagents may be sourced from any manufacturer ▲CRITICAL For media requiring the addition of acetosyringone, a note is provided at the bottom of the component list as a reminder that acetosyringone is added to the medium immediately before carrying out the procedure.
Reagents
Modular Golden Gate assembly of a T-DNA vector
Agrobacterium strain AGL1, EHA105 or GV3101 (Intact Genomics, cat no. 1083, 1084 or 1282)
BsaI-HFv2 (NEB cat. no. R3733S)
Bovine serum albumin (BSA; NEB, cat. no. B9000S)
Complementary oligonucleotides with overhangs (Sigma) (see Procedure 1, Step 2)
ddH2O, sterile
DH5-alpha (or similar) Escherichia coli competent cells (NEB, cat. no. C2987H)
1,4-Dithiothreitol (DTT; Sigma Aldrich, cat. no. 3483–12-3)
Esp3I (NEB, cat. no. R0734S; supplied with 10× reaction buffer)
Tryptone (Fisher Scientific, cat. no. BP1421–2)
NaCl (Fisher Scientific, cat. no. S271–3)
Yeast extract (Amresco, cat. no. J850–1KG)
Bacterial agar (IBI Scientific, cat. no. IB49172)
Glycerol (Sigma Aldrich, cat no. G5516–100ML)
PaqCI (NEB, cat. no. R0745S; supplied with 10× reaction buffer and PaqCI activator)
Plasmid miniprep kit (Qiagen, cat. no. 27104)
Plasmid-Safe (Bioscience Technologies, cat. no. EK3101K; supplied with 25 mM ATP)
Super Optimal Broth with catabolite repression (SOC) medium (Fisher, cat. no. 15544034)
Step 6 pTAG sgRNA vector backbones (Supplementary Table 1)
Step 15 cloning modules and B module vector backbone (Supplementary Table 2)
Step 19 cloning modules and T-DNA vector backbone (Supplementary Table 3)
T4 DNA ligase (NEB, cat. no. M0202S; supplied with 10× reaction buffer)
T4 polynucleotide kinase (PNK) (NEB, cat. no. M0201S; supplied with 10× PNK reaction buffer)
Spectinomycin (Gold Bio, cat no. S-140–5)
Carbenicillin (Gold Bio, cat no. C-103–25)
Kanamycin (Gold Bio, cat no. K-120–5)
Genamycin (Gold Bio, cat no. G-400–1)
Rifampicin (Gold Bio, cat no. R-120–1)
5-Bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-Gal; Gold Bio, cat. no. X4281C)
Dimethylformamide (DMF; Sigma Aldrich, cat. no. D4551–250ML)
Isopropyl-β-d-thiogalactoside (IPTG; Gold Bio, cat no. I2481C)
Direct delivery
Agrobacterium stock with cloned T-DNA vector (Procedure 1)
Appropriate antibiotics for Agrobacterium strain (Experimental design section)
Acetosyringone (Sigma Aldrich, cat. no. D134406)
Magnesium chloride (MgCl2·6H2O; Sigma Aldrich, cat no. M2670–500G)
2-(N-Morpholino) ethanesulfonic acid sodium salt (MES; Fisher Scientific, cat. no. BP300–100)
Cas9 or wild-type N. benthamiana seeds (Maher et al.6)
Potting soil
Tryptone (Fisher Scientific, cat. no. BP1421–2)
NaCl (Fisher Scientific, cat. no. S271–3)
Yeast extract (Amresco, cat. no. J850–1KG)
Bacterial agar (IBI Scientific, cat. no. IB49172)
Fast-TrACC
Murashige & Skoog basal medium with vitamins (PhytoTech Labs, cat. no. M519)
Sucrose (Fisher Scientific, cat. no. S5–3)
Gelzan (BidScientific, cat no. G024–1KG)
Ethanol, 70% (diluted from 100%)
Potassium phosphate (K2HPO4; Fisher Scientific, cat. no. P290–500)
Sodium phosphate (Na2HPO4; Fisher Scientific, cat. no. BP332–1)
Ammonium chloride (NH4Cl; Honeywell, cat. no. 09725–100G)
Magnesium sulfate heptahydrate (MgSO4·7H2O; Sigma Aldrich, cat. no. M1880–1KG)
Glucose (Sigma Aldrich, cat. no. G8270–1KG)
Iron sulfate, 10 mM; FeSO4 stock (PhytoTech Labs, cat. no. F318)
CaCl2·2H2O (Fisher Scientific, cat. no. C69–500)
KCl (Fisher Scientific, cat. no. P217–500)
KOH (Sigma Aldrich, cat no. 221473–25G)
Agrobacterium stock with cloned T-DNA vector (Procedure 1)
Commercial bleach (5.25% sodium hypochlorite)
ddH2O, sterile
2-(N-Morpholino) ethanesulfonic acid sodium salt (MES; Fisher Scientific, cat. no. BP300–100)
Indole-3-butyric Acid (IBA; Sigma Aldrich, cat no. I5386–1G)
Plant agar (Sigma Aldrich, cat. no. A1296–1KG)
Timentin (Gold Bio, cat. no. T-104–2)
Tween-20 (Sigma Aldrich, cat. no. P1379–25ML)
Acetosyringone (Sigma Aldrich, cat. no. D134406)
Tryptone (Fisher Scientific, cat. no. BP1421–2)
NaCl (Fisher Scientific, cat. no. S271–3)
Yeast extract (Amresco, cat. no. J850–1KG)
Bacterial agar (IBI Scientific, cat. no. IB49172)
Cas9 or wild-type N. benthamiana seeds (Maher et al.6)
Potting soil
Carbenicillin
Appropriate antibiotics for Agrobacterium strain (Experimental design section)
Equipment
Modular Golden Gate assembly of a T-DNA vector
−20 °C ice block (Fisher, cat. no. 355501)
Aluminum foil (Grainger, cat no. 16W483)
Ice bucket (Fisher, cat. no. 07–210-129)
Polypropylene microfuge tubes (1.7 ml; Millipore Sigma, cat. no. CLS3620–500EA)
Thermocycler
0.2 ml × 8 PCR strip tubes (MIDSCI, cat. no. PR-PCR28ACD)
DD
3-Inch plastic pots (Greenhouse Megastore, cat. no. CN-SQK 35)
28–32 gauge needle and syringe (Fisher, cat no. 328440)
Growth chamber: 22 °C, 16 h day/8 h night, 75–100 μmol m−2 s−1 of light, 40–60% humidity
Scalpel blade (#10) and holder (Fisher, cat. no. 08–914A; Fisher, cat. no. 12–000-163)
Plastic dome covering (Greenhouse Megastore, cat no. CNDOME)
Spectrophotometer
Forceps (15 cm; Fisher, cat. no. NC9131310)
Permanent marker
String tag
Fast-TrACC
Polypropylene microfuge tubes (1.7 ml; Millipore Sigma, cat. no. CLS3620–500EA)
6-Well plate (TrueLine, cat no. TR5000)
12-Well plate (TrueLine, cat no. TR5001)
Bead sterilizer (Fisher, cat. no. 14–955-340)
Conical centrifuge tube (15 ml; 50 ml)
Conical flask (rimless neck 125 ml; 500 ml)
Conical flask closure
Growth chamber: 22 °C, 16 h day/night, 50–150 μmol m−2 s−1 of light, 40% humidity
Forceps (15 cm; Fisher, cat. no. NC9131310)
Light rack incubator: 25 °C, 16 h day/night, 75–100 μmol m−2 s−1 of light, 40–60% humidity
Micropore tape (3M, cat. no. 1530–1)
3-Inch plastic pots (Greenhouse Megastore, cat. no. CN-SQK 35)
Scalpel blade (#10) and holder (Fisher, cat. no. 08–914A; Fisher, cat. no. 12–000-163)
Plastic dome covering (Greenhouse Megastore, cat no. CNDOME)
Spectrophotometer
General equipment
Autoclave
Flow hood
Vortex
0.22 μm filter (Midwest Scientific, cat no. TP99722)
Freezer: −80 °C, −20 °C, 4 °C
Cell spreader
Culture tubes (17 × 100 mm; Fisher, cat. no. 149569C)
Incubator: 28 °C, 37 °C
Shaking incubator: 28 °C, 37 °C
Micropipettes
Tabletop centrifuge
Water bath: 37 °C, 42 °C, 55 °C
Serological pipette controller
Serological pipettes (5 ml, 10 ml, 25 ml and 50 ml; MedSupply Partners, cat. no. 62–1005)
Petri plates (100 × 20 mm; USA Scientific, cat no. 5666–4161Q)
Reagent setup
1/2 MS liquid medium (1 l)
- Add the following reagents into 500 ml of ddH2O:
- 2.2 g of Murashige & Skoog basal medium with vitamins
- 5 g of sucrose
Fill to 1,000 ml with ddH2O
Adjust pH to 5.7 with KOH
Autoclave on liquid cycle for 30 min
Allow solution to cool in 55 °C in a water bath
Pour into 20 mm × 100 mm Petri plates
Store up to 1 month at 25 °C
1/2 MS solid medium (1 l)
- Add the following reagents into 500 ml of ddH2O:
- 2.2 g of Murashige & Skoog basal medium with vitamins
- 5 g of sucrose
Fill to 1,000 ml with ddH2O
Adjust pH to 5.7 with KOH
Add 3 g of Gelzan
Autoclave on liquid cycle for 30 min
Store up to 1 month at 4 °C
AB-MES medium (1 l)
- Add the following reagents into 500 ml of ddH2O:
- 3 g of potassium phosphate (K2HPO4)
- 1 g of sodium phosphate (Na2HPO4)
- 1 g of ammonium chloride (NH4Cl)
- 0.31 g of magnesium sulfate heptahydrate (MgSO4·7H2O)
- 10.66 g of MES
- 20 g of glucose
- 1 ml of 2 M potassium chloride (KCl stock)
- 100 μl of 1 M calcium chloride (CaCl2 stock)
- 1 ml of 10 mM iron sulfate (FeSO4 stock)
Fill to 1,000 ml with ddH2O
Adjust pH to 5.7 with KOH
Autoclave on liquid cycle for 30 min
Store up to 6 months at 25 °C. ▲CRITICAL Prior to use of AB-MES medium in Procedure 3, Step 16, add appropriate volume of 100 mM acetosyringone stock for a final concentration of 200 μM.
Acetosyringone stock (100 mM, 10 ml)
Add 0.196 g of acetosyringone into 5 ml of methanol
Fill to 10 ml with ddH2O
Filter-sterilize with a 0.22 μm filter
Divide into 1 ml aliquots
Store up to 6 months at 20 °C
Agrobacterium activation medium (50 ml)
- Add the following reagents to 50 ml of sterile LB medium:
- 1 ml of 0.5 M MES stock solution
- 10 μl of 100 mM acetosyringone stock (20 μM final)
Appropriate antibiotics for Agrobacterium strain (Experimental design section).
Use solution immediately in Procedure 2, Step 9
Antibiotic stocks (50 mg ml−1, 5 ml)
▲CRITICAL The following recipe works for antibiotics used at a concentration of 50 mg ml−1. These include spectinomycin, kanamycin, carbenicillin and gentamycin.
Add 50 mg of anhydrous antibiotic to 3 ml of ddH2O
Mix by vortexing
Fill to 5 ml with ddH2O
Filter sterilize with a 0.22 μm filter
Divide into 1 ml aliquots
Store up to 6 months at 20 °C
Calcium chloride (CaCl2) stock solution (1 M, 50 ml)
Add 7.4 g of CaCl2·2H2O into 30 ml of ddH2O
Fill to 50 ml with ddH2O
Filter-sterilize with a 0.22 μm filter
Store up to 12 months at 4 °C
Co-cultivation medium (100 ml)
- In a flow hood, add the following reagents to a sterile Erlenmeyer flask:
- 50 ml of 1/2 MS liquid medium
- 50 ml of AB-MES medium
- 200 μl of 100 mM acetosyringone stock
Use solution immediately in Procedure 3, Steps 21 and 22. ▲CRITICAL Prior to use of co-cultivation medium in Procedure 3, Step 21 and 22, add an appropriate volume of 100 mM acetosyringone stock for a final concentration of 200 μM.
Glycerol stock solution (50%, 100 ml)
Add 50 ml of glycerol to 50 ml of ddH2O
Autoclave on liquid cycle for 20 min
Store up to 1 year at 25 °C
Injection medium (1 l)
- Add the following reagents to 500 ml of ddH2O:
- 2.03 g of magnesium chloride (MgCl2·6H2O) (final 10 mM)
- 2.13 g of MES (final 10 mM)
Fill to 1,000 ml with ddH2O
Filter-sterilize with a 0.22 μm filter
Store up to 1 year at room temperature at (25 °C). ▲CRITICAL Prior to use of injection medium in Procedure 2, Step 12, add appropriate volume of 100 mM acetosyringone stock for a final concentration of 200 μM.
LB agar medium (1 l)
- Add the following reagents to 500 ml of ddH2O:
- 10 g of tryptone
- 5 g of NaCl
- 5 g of yeast extract
Fill to 1,000 ml with ddH2O
Add 15 g of bacterial agar
Autoclave on liquid cycle for 30 min
Allow solution to cool to 55 °C in a water bath
Add appropriate antibiotic stock
Pour into 20 mm × 100 mm Petri plates
Store up to 1 month at 4 °C. ▲CRITICAL For plates that contain X-Gal and IPTG (Procedure 1, Step 9), pipette 30 μl of each stock solution onto solid plates, spread with a cell spreader and allow to dry.
LB liquid medium (1 l)
- Add the following reagents to 500 ml of ddH2O:
- 10 g of tryptone
- 5 g of NaCl
- 5 g of yeast extract
Fill to 1,000 ml with ddH2O
Autoclave on liquid cycle for 30 min
Store up to 1 year at 25 °C
MES stock solution (0.5 M, 100 ml)
Add 10.5 g of MES into 80 ml ddH2O
Adjust the pH to 5.6 with NaOH
Fill to 100 ml with ddH2O
Filter-sterilize with a 0.22 μm filter
Store up to 1 year at 25 °C
Potassium chloride (KCl) stock solution (2 M, 50 ml)
Add 7.5 g of KCl into 30 ml ddH2O
Fill to 50 ml with ddH2O
Autoclave on liquid cycle for 30 min
Store up to 1 year at 25 °C
Rooting medium23 (1 l)
- Add the following reagents into 500 ml of ddH2O:
- 2.15 g of Murashige & Skoog basal medium with vitamins
- 30 g of sucrose
Fill to 1,000 ml with ddH2O
Adjust the pH to 5.2 with KOH
Add 8 g of plant agar
Autoclave on liquid cycle for 30 min
Allow solution to cool to 55 °C in a water bath
- In a sterile hood, add the following reagents:
- 500 μl of 1 mg ml−1 IBA
- 2 ml of 50 mg ml−1 carbenicillin
- 1 ml of 200 mg ml−1 timentin
Pour into 20 mm × 100 mm Petri plates
Store up to 1 month at 4 °C
Timentin stock (200 mM, 5 ml)
Add 0.66 g of timentin to 3 ml of ddH2O
Fill to 5 ml with ddH2O
Filter-sterilize with a 0.22 μm filter
Divide into 1 ml aliquots
Store up to 2 months at −20 °C
X-Gal stock (40 mg ml−1, 5 ml)
Add 200 mg of X-Gal to 5 ml of DMF
Vortex to dissolve
Divide into 1 ml aliquots in polypropylene microfuge tubes
Wrap tubes in aluminum foil to prevent degradation by light
Store up to 6 months at −20 °C
IPTG stock (100 mM, 10 ml)
Add 0.238 g of IPTG to 5 ml of ddH2O
Fill to 10 ml with ddH2O
Filter sterilize with a 0.22 μm filter
Divide into 1 ml aliquots
Store up to 1 year at −20 °C, protected from light
Procedure
Procedure 1: modular Golden Gate assembly of a T-DNA vector
▲CRITICAL The following steps in Procedure 1 describe the cloning of Cas9 sgRNA-tRNA array vectors. This array cloning system allows the assembly of up to six different guide RNA sequences from a single polymerase II promoter in even-numbered (two, four or six) sets of sgRNAs. Steps 1–6 will need to be repeated for each sgRNA. To determine which plasmid backbones to use in Step 6, see Supplementary Table 1. For further information on sgRNA-tRNA arrays, see Experimental design section.
Design sgRNA(s) for target gene ● Timing 1 day
-
1
Using bioinformatics software, such as Geneious, Benchling or other openly available online resources, design Cas9 sgRNA spacers for a specific gene target(s).
-
2
Synthesize the sgRNA as a pair of oligos, complementary for the spacer sequence and prepended with the following 5′ Golden Gate cloning overhangs:
5′ – TGCA – XXXXXXXXXXXXXXXXXXXX
XXXXXXXXXXXXXXXXXXXX – CAAA – 5′
▲CRITICAL STEP sgRNA spacers should not contain recognition sequences for type IIs restriction enzymes BsaI, Esp3I or PaqCI to prevent complications in future cloning steps.
-
3
Order these oligos from any DNA synthesis provider at a scale of 0.025 μM, requesting desalting purification and no modifications.
▲CRITICALSTEP Oligos can be ordered with 5′ phosphates. If this option is selected, anneal oligos without phosphorylation reaction in Step 4.
Cloning synthesized oligos into tRNA MoClo compatible vectors ● Timing 3 d
-
4Phosphorylate and anneal synthesized oligos by setting up and running the following reaction (Fig. 2a):
Phosphorylation reactionmMixture: Amount (μl) Final concentration Top-strand sgRNA oligonucleotide (100 μM) 3 10 μM Bottom-strand sgRNA oligonucleotide (100 μM) 3 10 μM T4 DNA ligase buffer (10×) 3 1× T4 polynucleotide kinase 2 H2O 19 Total 30 Thermocycler settings Cycle number Temperature Time Ramp setting 1 37 °C 1 h 2 95 °C 5 min 3 85 °C 2 °C 4 25 °C −0.1 °C 5 4 °C Hold -
5
Dilute the reaction 1:24 with ddH2O.
■PAUSE POINT Annealed oligos can be stored at −20 °C for several months.
-
6Prepare separate Golden Gate reactions with appropriate pTAG sgRNA backbones for each pair of oligos annealed in Step 4 (Supplementary Table 1) using the following reagents and conditions (Fig. 2a):
Golden Gate reaction mixture Amount (μl) Final concentration pTAG sgRNA backbone (Supplementary Table 1) 150 ng 1:24 annealed oligos (−0.4 μM, Step 5) 1 −0.02 μM Esp3I 0.5 T4 DNA ligase buffer (10×) 2 1× DTT (10 mM) Fill to 20 Total 20 Thermocycler settings Cycle number Temperature Time 1 37 °C 5 min 2 16 °C 10 min 3 37 °C 15 min 4 80 °C 5 min 5 10 °C Hold ■PAUSE POINT Golden Gate reaction to create pTAG vector backbone can be stored at −20 °C. Circular DNA should be stable for several months.
-
7
Following the manufacturer’s instructions, transform ~5 μl of each Golden Gate reaction into ~50 μl of competent DH5-alpha (or similar) E. coli cells.
-
8
Add 150 μl of SOC to transformed cells and shake for 1 h at 250 rpm in a 37 °C shaking incubator.
-
9
Plate ~50 μl of cells on prepared LB plates containing 50 mg l−1 spectinomycin, X-Gal and IPTG (Reagents setup section).
▲CRITICALSTEP Immediately before use, add 30 μL of X-Gal and IPTG to plates of solid medium and spread using sterile spreading device.
-
10
Place the plates in a 37 °C incubator for 16 h.
-
11
Once colonies are visible, pick two or three white colonies for separate 5 ml LB liquid cultures containing 50 mg l−1 spectinomycin.
■PAUSE POINT E. coli plates with visible colonies can be stored at 4 °C for several weeks.
? TROUBLESHOOTING
-
12
Shake 5 ml cultures for 16 h at 250 rpm in a 37 °C shaking incubator.
? TROUBLESHOOTING
-
13
Following the manufacturer’s instructions, purify pTAG sgRNA vectors using a plasmid miniprep kit.
■PAUSE POINT Purified plasmid can be stored at −20 °C for years.
-
14
Sequence the purified vectors to verify the integrity of pTAG sgRNA vectors before proceeding to Step 15 using primers listed in Supplementary Table 6.
? TROUBLESHOOTING
Construction of module B – guide ● Timing 3 d
-
15
Select a promoter module, terminator module and B module vector backbone using Supplementary Table 2.
▲CRITICAL STEP Promoter and terminator vectors in this assembly step are compatible with the Golden Gate-based MoClo cloning system. Promoter and terminator vectors listed in Supplementary Table 2 may be interchanged with certain vectors from this system9. For source kits see: addgene.org/cloning/moclo.
-
16Set up a Golden Gate cloning reaction using the following reagents and conditions (Fig. 2b):
Golden Gate reaction mixture Amount (μl) Final concentration Promoter module vector (Supplementary Table 2) 150 ng Assembled sgRNA module vectors (Steps 1–14) 150 ng per vector Terminator module vector (Supplementary Table 2) 150 ng B module vector backbone (Supplementary Table 2) 75 ng BsaI-HFV2 1 T4 DNA ligase 1 T4 DNA ligase buffer (10×) 2 1× BSA (10×) 2 1× DTT (10 mM) Fill to 20 Total 20 Thermocycler settings Cycle number Temperature Time 1 37 °C 5 min 2 16 °C 10 min 3 Go to Cycle number 1 (10–40×) 4 37 °C 15 min 5 80 °C 5 min 6 10 °C Hold -
17
Treat reaction with 1 μl of Plasmid-Safe ATP-dependent DNase and 1 μl of 25 mM ATP. Incubate at 37 °C for 1 h.
■PAUSE POINT Golden Gate reaction to create the B module backbone can be stored at −20 °C. Circular DNA should be stable for several months.
-
18
Repeat Steps 7–14, using LB plates containing 50 mg l−1 carbenicillin in Step 9 and media containing 50 mg l−1 carbenicillin in Step 11. In Steps 13 and 14, purify the assembled B module vector containing the assembled guide expression cassette and sequence the purified vectors to verify the integrity of the B module assembly before proceeding to Step 19 using primers listed in Supplementary Table 6.
■PAUSE POINT E. coli plates with visible colonies can be stored at 4 °C for several weeks.
▲CRITICAL STEP The sequencing primers used depend on the number of spacer sequences cloned and the promoter and terminator chosen. Given the repetitive nature of the sgRNA array, it is recommended to confirm the entire sgRNA array by DNA sequencing.
▲CRITICAL STEP Depending on the melting temperature (Tm) of the oligos used to make the first and last sgRNA in the array, the first ‘sense’ oligo and last ‘antisense’ oligo can be used to confirm the sequence of the sgRNA array.
? TROUBLESHOOTING
Assembly of T-DNA vector ● Timing 3 d
-
19
Select module vectors A, C′ and D and Replicon T-DNA backbone from Supplementary Table 3.
▲CRITICAL STEP Depending on the downstream transformation protocol chosen (DD or Fast-TrACC), some regulator modules may not be desired in the final replicon T-DNA vector assembly (see Experimental design section for further discussion of the choice of DRs). If this is the case, ‘empty 0000’ vectors may be selected for the Golden Gate assembly in Step 20 (Supplementary Table 3).
-
20Using the assembled B module (guides) from Steps 15–18, set up a Golden Gate cloning reaction using the following reagents and conditions (Fig. 2c):
Golden Gate reaction mixture Amount (μl) Final concentration Module A – reporter or gene editing reagent (Supplementary Table 3) 150 ng Module B – guides (Steps 15–18) 150 ng Module C’ – regulator (Supplementary Table 3) 150 ng Module D – regulator (Supplementary Table 3) 150 ng Backbone – replicon T-DNA (Supplementary Table 3) 75 ng BSA (10×) 0.5 0.5× DTT (10 mM) 0.5 0.5× T4 DNA ligase buffer (10×) 2 1× T4 DNA ligase 1 PaqCI 1 PaqCI activator 0.4 H2O Fill to 20 Total 20 Thermocycler settings Cycle number Temperature Time 1 37 °C 5 min 2 16 °C 10 min 3 Go to Cycle number 1 (10–40×) 4 37 °C 15 min 5 80 °C 5 min 6 10 °C Hold ■PAUSE POINT Golden Gate reaction to create the T-DNA backbone can be stored at −20 °C. Circular DNA should be stable for several months.
-
21
Repeat Steps 7–14, using LB plates containing 50 mg l−1 kanamycin in Step 9 and media containing 50 mg l−1 kanamycin in Step 11. In Steps 13 and 14, purify the assembled replicon T-DNA vector containing the assembled A, B, C′ and D modules. Sequence the purified vectors to verify the integrity of the assembly before proceeding to Step 22.
▲CRITICAL STEP Sequencing primers depend greatly on which modules are used. Use sequencing primers that allow verification of the ligation junctions between the vector backbone and all the individual modules. Primers should be designed according to the promoters and terminators used, ensuring that no primers bind more than one location on a given vector. For primer design, free software, such as Benchling or Primer3, can be used.
? TROUBLESHOOTING
Transformation into Agrobacterium ● Timing 3 d
-
22
Remove competent Agrobacterium cells from −80 °C freezer and thaw on ice.
-
23
Following the manufacturer’s instructions, transform verified replicon T-DNA vector from Step 21 into competent Agrobacterium cells and follow recommended recovery steps.
-
24
Plate 50–100 μl of cells on LB plates containing appropriate antibiotics for selected Agrobacterium strain.
▲CRITICAL STEP Agrobacterium strains AGL1 and EHA105 require 50 mg l−1 kanamycin and 10 mg l−1 rifampicin for selection. GV3101 requires 50 mg l−1 kanamycin, 50 mg l−1 gentamycin and 10 mg l−1 rifampicin.
-
25
Place plates in a 28 °C incubator for 2 d.
-
26
After colonies are visible, pick a single colony for a 5 ml LB liquid culture containing appropriate antibiotics for selected Agrobacterium strain.
■PAUSE POINT Agrobacterium plates with visible colonies can be stored at 25 °C for at least 1 week.
? TROUBLESHOOTING
-
27
Shake 5 ml culture tubes for 20–24 h at 28 °C. Use this culture to create a permanent glycerol stock by mixing at a 1:1 ratio with prepared glycerol stock solution. Store glycerol stock at −80 °C.
▲CRITICAL STEP The Agrobacterium stock containing the replicon T-DNA can be used in either DD (Procedure 2) or Fast-TrACC (Procedure 3). Depending on the desired plant transformation method (see Experimental design section for further details), proceed to the appropriate section.
Procedure 2: DD
Plant selection and development ● Timing 4–8 weeks
-
1
Plant three or four Cas9 or wild-type N. benthamiana seeds per 3-inch plastic pot filled with dampened potting soil.
-
2
Place pots in the growth chamber and cover with a plastic dome for 1 week to increase humidity and improve germination.
-
3
Once seedlings are visible, use forceps to eliminate all but one plant per pot.
▲CRITICAL STEP Researchers should use at least eight plants per experiment.
-
4
Place seedlings in the growth chamber for 4–8 weeks to allow for plant growth and the initiation of axillary meristems at leaf axils (Fig. 3a).
▲CRITICAL STEP While younger plant tissues have higher plasticity to transdifferentiate and form de novo meristems, plants with visible axillary meristems and increased lateral growth will be easier to use in DD experiments.
-
5
Once axillary meristems are visible, proceed to Step 6.
Fig. 3 |. DD procedure and sample outcomes.

a, Individual N. benthamiana plants are grown in soil in at least 3-inch pots. Plants are sufficiently mature when axillary shoots have developed. b, All existing meristems (shoot apical and axillary meristems) are removed using a scalpel. c, After trimming, plants have two to three nodes and a few supporting leaves with no observable shoot meristems. d,e, Agrobacterium cultures are delivered using a syringe to leaf axils and all wound sites (shoot apices and sites of axillary meristems, white arrows in e). f–h, Examples of shoots that emerge after a 20 day culling period. Among these are wild-type (f) and edited (g) shoots. Some shoots may have morphological abnormalities due to constitutive overexpression of the DRs (h). Reproduced with permission from ref. 6, Springer Nature Limited.
Agrobacterium preparation for DD ● Timing 3 d
-
6
Remove the Agrobacterium stock containing assembled replicon T-DNA from −80 °C storage (from Step 27, Procedure 1).
-
7
Scrape surface of the frozen Agrobacterium glycerol stock with a sterile utensil and streak onto an LB agar plate containing appropriate antibiotics for selected Agrobacterium strain. Return Agrobacterium stock to −80 °C freezer.
▲CRITICALSTEP To maintain cell stock viability, it is important that bacterial glycerol stocks do not thaw during this process.
-
8
Place the Agrobacterium plate in a 28 °C incubator for 1–2 d.
▲CRITICAL STEP Growth will be noticeable after 24 h; however, a 2 day incubation period is needed to obtain colonies of adequate size.
-
9
Once visible, pick a single Agrobacterium colony from the plate and transfer to a 5 ml culture tube containing Agrobacterium activation medium.
-
10
Place the culture tube in a 28 °C shaking incubator set to ~250 rpm for 16 h.
▲CRITICAL STEP Overnight growth in LB containing MES and acetosyringone at low concentrations activates expression of Agrobacterium vir genes.
-
11
Centrifuge at 6,000g for 10 min at 25 °C to pellet the Agrobacterium cells, and discard the supernatant.
-
12
Resuspend pellet in 3 ml of injection medium, and read cell density using a spectrophotometer. Adjust the cell density to OD600 = 0.2 with injection medium containing acetosyringone.
▲CRITICALSTEP For experiments requiring the co-delivery of two different Agrobacterium strains, mix the solutions together in 1:1 ratio; for three strains, mix in 1:1:1 ratio.
-
13
Incubate Agrobacterium at 25 °C (room temperature) for 2–4 h. This suspension will be used in Step 15. Immediately proceed to Step 14 to prepare N. benthamiana plants for DD.
Plant preparation and DD of Agrobacterium ● Timing ~2 h
-
14
Referring to Fig. 3b,c, use a scalpel or single-edged razor blade to remove all apical and axillary meristems from mature N. benthamiana plants.
▲CRITICAL STEP It is important to retain supporting leaves associated with delivery sites at each leaf axil to facilitate meristem formation. Remove other unnecessary tissues to reduce background meristem formation.
-
15
Using a syringe with a 28–32 gauge needle, draw up ~1 ml of the adjusted Agrobacterium injection medium suspension from Step 13 and carefully inject into nodal tissues at leaf axils and wound sites outlined in Fig. 3d,e.
▲CRITICAL STEP To keep track of the inoculation sites, leaf stems may be marked at delivery sites with a permanent marker or string tag.
-
16
Place injected plants in the growth chamber, and proceed to Step 17 for instructions on plant maintenance.
Plant maintenance for DD ● Timing 20 d post inoculation
-
17
At 2–3 d after inoculation in Step 16, use a scalpel or single-edged blade to remove and discard all newly formed meristems from the entire plant, taking care not to remove plant tissue initially inoculated with Agrobacterium. Repeat this process every 2–3 d up until 20 d post inoculation.
▲CRITICALSTEP Continuous culling of early shoots ensures removal of primordial meristems that were missed in initial trimming and reduces spontaneous background shooting, which has been found to predominantly occur during this time period.
? TROUBLESHOOTING
De novo shoot formation for DD ● Timing 2–4 weeks
-
18
After 20 d, discontinue shoot removal at inoculation sites to allow for DR-induced meristems to form. Formation of induced meristems should be observed 38–48 d post inoculation.
▲CRITICALSTEP It is recommended to continue culling shoots that emerge at noninoculation sites to prevent a spontaneous shoot from noninoculated tissues achieving apical dominance.
? TROUBLESHOOTING
Screening de novo shoots for genetic modifications in DD ● Timing variable
-
19
De novo tissues should be screened for genetic modifications of interest once leaves have matured; leaves may be sampled by making a leaf punch.
▲CRITICAL STEP Transgenic tissues constitutively overexpressing DRs may have a variety of phenotypes (Fig. 3f–h). Some tissues may be unable to support induction of viable vegetative or floral shoots.
-
20
To isolate gDNA, use a cetyltrimethyl ammonium bromide (CTAB) protocol26 or a similar extraction method.
-
21
Using extracted gDNA from leaf tissue, analysis of edits can be carried out using the Tracking of Indels by DEcomposition (TIDE) method27.
? TROUBLESHOOTING
Procedure 3: Fast-TrACC
▲CRITICAL All steps in the following procedure should be carried out in a sterile flow hood. Exceptions include centrifugation and incubation of closed, sterile Petri dishes.
Seed surface sterilization and germination for Fast-TrACC ● Timing 4–5 d
▲CRITICAL Steps 1–9 should be carried out in parallel with the Agrobacterium preparation process of this procedure (Steps 10–18, Procedure 3).
-
1
Measure out ~40 mg of Cas9 or wild-type N. benthamiana seeds and place in a 1.7 ml microfuge tube.
-
2
Inside a sterile flow hood, pipette 1 ml of 70% ethanol into the tube with the seeds and mix by gently inverting two or three times.
-
3
Wait 2–3 min before carefully removing the ethanol with a pipette and discarding.
-
4
Pipette 1 ml of a 1:20 mixture of commercial bleach (5.25% sodium hypochlorite) and sterile ddH2O with two or three drops of Tween-20 into the seed tube, and mix by gently inverting two or three times.
-
5
Wait 10–15 min to allow the bleach solution to completely surface-sterilize the seeds, periodically inverting tubes.
-
6
Using a pipette, carefully remove the bleach solution without disturbing the seeds and discard.
-
7
Wash seeds three or four times with 1 ml of sterile ddH2O to remove residual bleach, each time discarding the liquid.
-
8
After the final wash, resuspend the seeds in 1/2 MS liquid medium and evenly distribute 10–15 seeds/well in a six-well plate using a pipette (Fig. 4a).
▲CRITICAL STEP Alternatively, the seeds can quickly be poured into a single well in the six-well plate and evenly distributed using a pipette.
-
9
Seal the plate with micropore tape and place in a light rack incubator for 4–5 d to allow seeds to germinate.
Fig. 4 |. Fast-TrACC procedure and sample outcomes.

a, N. benthamiana seedlings are germinated in sterile six-well plates. b, Once the cotyledons emerge, as shown in this single-well close-up, the liquid medium is removed and replaced with the treated Agrobacterium culture. c, After 2 d of co-culture, the seedlings are rinsed in water and individual seedlings are transferred to wells in a 12-well plate. d, After about 20 d, green growths begin to form on the seedlings (white arrow). These growths can be maintained on the seedling (e) or removed and transferred to solid 1/2 MS medium (f). Shooting has been observed to occur directly on seedlings (e, white arrow); however, most de novo growths continue to increase in mass without shooting (e, black arrow). f, Transfer of de novo growths to 1/2 MS medium can stimulate shoot development. g, Upon shooting, the growths can be transferred to root-inducing medium to promote root formation. h, Finally, the plantlets with established root systems can be transferred to soil for further growth.
Agrobacterium preparation for Fast-TrACC ● Timing 4 d
▲CRITICAL The following steps describe the treatment of a single six-well plate of germinating N. benthamiana seeds. For transformations that require more than a single six-well plate, scale the volumes accordingly.
-
10
Remove the Agrobacterium stock containing assembled replicon T-DNA from the −80 °C freezer (from Step 27, Procedure 1).
-
11
Streak out the Agrobacterium stock onto an LB agar plate containing appropriate antibiotics for selected Agrobacterium strain.
-
12
Place the plate in a 28 °C incubator for 1–2 d.
▲CRITICALSTEP Growth will begin to be noticeable after 24 h; however, a 2 day incubation period is needed to obtain colonies of adequate size.
-
13
Once visible, pick a single Agrobacterium colony from the plate and transfer to 5 ml of liquid LB medium containing appropriate antibiotics.
-
14
Place the culture tube in a 28 °C shaking incubator set to ~250 rpm for 20–24 h.
-
15
Centrifuge at 6,000g for 10 min at 25 °C to pellet the Agrobacterium cells, and discard the supernatant.
-
16
In a sterile flow hood, resuspend the pellet in 3 ml of AB-MES and mix by briefly vortexing.
-
17
Using a serological pipette, transfer 12 ml of fresh AB-MES to a sterile conical flask. With the 3 ml Agrobacterium resuspension from Step 16, adjust the cell density of AB-MES to OD600 = 0.2.
-
18
Place the AB-MES culture flask in a 28 °C shaking incubator set to ~250 rpm for 12–16 h.
Agrobacterium co-cultivation for Fast-TrACC ● Timing 2 d
-
19
Using a serological pipette, remove 12 ml of the AB-MES culture and place into a sterile 15 ml conical centrifuge tube.
-
20
Centrifuge at 6,000g for 10 min at 25 °C to pellet the Agrobacterium cells, and discard the supernatant.
-
21
Resuspend pellet in 3 ml of co-cultivation medium. Place Agrobacterium resuspension in a tube rack. It will be used in Step 23.
-
22
Using a serological pipette, transfer 25 ml of co-cultivation medium into a sterile 50 ml conical centrifuge tube.
-
23
Using a pipette, with the Agrobacterium suspension from Step 21, adjust the cell density of the 25 ml volume of co-cultivation medium to OD600 = 0.10–0.18. Place the adjusted Agrobacterium suspension in a tube rack. It will be used in Step 26.
▲CRITICALSTEP For experiments requiring the co-delivery of two different Agrobacterium strains, mix the solutions together in 1:1 ratio; for three strains, mix together in 1:1:1 ratio.
-
24
Remove the six-well plate containing germinated N. benthamiana from the light rack incubator (see Steps 1–9 of this procedure).
▲CRITICAL STEP The emerging N. benthamiana cotyledons should be visible prior to co-cultivation. See Fig. 4b for reference.
-
25
Using a serological pipette, remove the 1/2 MS medium from each well, leaving only the germinated seeds.
▲CRITICALSTEP It is easiest to tilt the six-well plate to pool the 1/2 MS for removal. Remember to tilt towards the flow hood filter to avoid possible contamination.
-
26
Using a serological pipette, transfer 3 ml of Agrobacterium co-culture suspension from Step 23 into each well of the six-well plate containing N. benthamiana seedlings.
-
27
Properly label the plate with the date and time of infection, seal with micropore tape and place in a light rack incubator. After 48 h of infection, proceed to Step 28.
? TROUBLESHOOTING
Transfer to growth development plates (Fast-TrACC) ● Timing 1 day (hands-on), 14 d (growth)
-
28
Carefully remove the six-well plate with Agrobacterium co-cultivation from the light rack incubator and place in a flow hood. This will be used in Step 31.
-
29
Using a serological pipette, transfer 2 ml of 1/2 MS + 200 μM timentin into each well of a 12-well plate. Cover and set aside. This will be used in Step 32.
▲CRITICAL STEP The required volume of 1/2 MS + 200 μM timentin will vary depending on the number of seedlings infected in Step 26. In general, 30 ml of 1/2 MS + 200 μM timentin should be prepared for every 12 seedlings infected.
-
30
Place a sterile Petri dish in the flow hood and half fill with sterile ddH2O.
-
31
Using sterile forceps, move seedlings from the six-well co-culture plate from Step 28 to the Petri dish with sterile ddH2O from Step 30 to wash off the Agrobacterium; completely submerge the cotyledons (Fig. 4c).
▲CRITICAL STEP It is not necessary to do each seedling individually. To increase the speed of transfer, multiple seedlings can be moved into the sterile ddH2O simultaneously. However, it is important to ensure that each seedling is completely washed to reduce Agrobacterium contamination.
-
32
Using sterile forceps, move individual seedlings from the ddH2O into single wells on the 12-well plate containing 1/2 MS + 200 μM timentin from Step 29 (Fig. 4c).
-
33
Seal the 12-well plate with micropore tape and place in a light rack incubator for 14 d.
Seedling maintenance ● Timing 1 day (hands-on), 6 d (growth)
-
34
Remove the 12-well plate from the light rack incubator and place in a sterile hood.
-
35
Using a sterile scalpel, trim back the newly emerging true leaves by making a transverse cut above the cotyledons (Fig. 4d).
-
36
Using a serological pipette, refill each well on the 12-well plate with fresh 1/2 MS + 200 μM timentin.
-
37
Reseal the 12-well plate with micropore tape and place in a light rack incubator for 6 d.
-
38
After shoot-like growths are visible on leaf tissue of transformed seedlings (Fig. 4e), proceed to Step 39 for shoot formation.
▲CRITICALSTEP Meristems may ectopically form from growths on seedling plants (Fig. 4e). These meristems may be transferred to rooting medium (Steps 47–49).
■PAUSE POINT Seedlings can be kept in 12-well plates for several days before transfer to solid media, but it is important to monitor levels of 1/2 MS in each well to keep plants healthy.
? TROUBLESHOOTING
Shoot formation ● Timing 2 d to 2 weeks
-
39
Remove the 12-well plate from the light rack incubator and place in a sterile hood.
-
40
Using a sterile scalpel and forceps, remove each growth from seedling leaf tissue and transfer to 1/2 MS solid medium plates.
-
41
Seal each plate with micropore tape and place in a light rack incubator for 2 weeks.
-
42
After 2 weeks, remove the 12-well plate from the light rack incubator and place in a sterile hood.
-
43
If a shoot continues to develop (Fig. 4f), transfer the shoot to rooting medium (Step 47). If the growths have no visible shoots, proceed to growth cultures (Steps 44–46).
▲CRITICAL STEP The timing of shoot formation is highly variable, and it will only occur on a subset of plant growths. All shoots should be transferred to rooting medium. The remainder of the growths are transferred to new plates.
Growth cultures ● Timing every 2 weeks
-
44
Using sterile forceps, transfer growth tissue as a whole to a fresh 1/2 MS solid medium plate.
-
45
Reseal plates with micropore tape and place in a light rack incubator for 2 weeks.
-
46
Repeat steps 44 and 45 every 2–3 weeks to replenish nutrients and perpetuate the growths.
▲CRITICAL STEP During this process, it is possible for more shoots to develop. If new shoots are identified, proceed to Step 47.
Root induction ● Timing 2–3 weeks
-
47
Remove plates with newly formed shoots from the light rack incubator and place in a sterile hood before removing the micropore tape.
-
48
Using a sterile scalpel and forceps, carefully transfer individual shoots to a rooting medium plate.
▲CRITICAL STEP For shoot removal, it is suggested to make a transverse cut near the base of the shoot.
▲CRITICAL STEP To allow adequate growth, leave at least ~1 cm of space between each shoot.
-
49
Seal plate with micropore tape and place in a light rack incubator for 2–3 weeks.
■PAUSE POINT After roots have formed, the plants can be kept on plates for several weeks before transfer to soil (Fig. 4g).
? TROUBLESHOOTING
Soil transfer of T0 plants ● Timing 1 d
-
50
Using forceps, remove regenerated T0 plants from rooting medium.
-
51
Rinse roots under running water to remove residual medium attached to the plant.
-
52
Carefully transfer T0 plants into 3-inch plastic pots filled with soil, with the base of the plant ~1 cm below the surface (Fig. 4h).
-
53
Transfer plants into a growth chamber, and cover the flat with a humidity dome for 5 d.
-
54
After 5 d, remove the dome and allow plants to proceed through development.
? TROUBLESHOOTING
Screening de novo shoots for genetic modifications in Fast-TrACC ● Timing variable
-
55
Regenerated plants should be screened for genetic modifications of interest once leaves have matured; leaves may be sampled by making a leaf punch.
-
56
To isolate gDNA from leaf tissue, use CTAB or a similar extraction method26.
-
57
Using extracted gDNA, analysis of targeted edits can be carried out using TIDE27.
Troubleshooting
Experiments described in Maher et al. used gene cassettes that constitutively expressed DR genes; however, pleiotropic effects due to DR overexpression may be deleterious to continued plant growth6. These negative effects may be circumvented in several ways. Expression control systems may be employed that constrain DR expression to a limited temporal or spatial window as demonstrated with recent applications of Wus2 and Bbm in monocots4. Further, experimental approaches may be taken that avoid downstream toxicity of DR expression by delivering DRs in trans on separate vectors25. This ‘altruistic’ approach has the benefit of creating a microenvironment for organogenesis to occur without integration of the T-DNA. Troubleshooting advice for Procedures 1, 2 and 3 can be found in Tables 1, 2 and 3, respectively.
Table 1 |.
Troubleshooting for Procedure 1: molecular cloning
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
|
| |||
| 11 | Cannot obtain white E. coli colonies with sgRNA array of interest | IPTG and XGal reagents were not added to plates | Refer to ‘Critical’ under LB agar medium in the Reagents setup section for instruction |
| 12 | No visible growth of E. coli in liquid culture | Incorrect antibiotic was used in setting up the overnight culture | Create a new overnight culture using E. coli plates from Step 11, ensuring the correct antibiotic is used for selection |
| 14 | Sequencing of inserted gene or sgRNA array shows mutations at GG overlap junctions | Golden Gate reaction likely did not occur to completion | Ensure that T4 DNA ligase buffer is fresh (contains sufficient DTT and ATP); Add more cycles to GG reaction (30–50) in Step 6 |
| 18 | No visible colonies after 16 h of growth at 37 °C | E. coli have been plated using incorrect antibiotic selection | If transformed E. coli cells from Step 18 were stored, replate cells on LB plates containing the appropriate antibiotics. If recovered cells were not stored, retransform Golden Gate reaction from Step 17 |
| Ligase activity may be compromised | Repeat Step 16 with fresh T4 DNA ligase and buffer | ||
| A component from the Golden Gate reaction mixture on Step 16 was omitted | Repeat Step 16, ensuring that each component is added to the reaction mixture | ||
| No visible growth of E. coli in liquid culture | Incorrect antibiotic was used in setting up the overnight culture | Create a new overnight culture using E. coli plates from Step 18, ensuring the correct antibiotic is used for selection | |
| Incorrect antibiotic was used when plating the recovered cells in Step 18 | Precursor backbones can be transformed and selected on solid LB medium if spectinomycin is used. If possible, replate stored cells from Step 18 on medium containing appropriate antibiotics, otherwise retransform Golden Gate reactions from Step 17 | ||
| Sequencing of inserted sequences shows mutations at GG overlap junctions | Golden Gate reaction likely did not occur to completion | Ensure that T4 DNA ligase buffer is fresh (contains sufficient DTT and ATP); Add more cycles to Golden Gate reaction (30–50) in Step 16 | |
| Sequencing of inserted genes of interest repeatedly reveals mutations in the reading frame | Some gene products may express in E. coli and be toxic. This is often observed as repeated novel single-nucleotide polymorphsisms in the reading frame | Consider adding an intron into the coding sequence | |
| If gene product was synthesized by vendor, it may be a mixed population | Contact vendor for resynthesis or screen multiple colonies to find a correct clone | ||
| 21 | No visible colonies after 16 h of growth at 37 °C | E. coli have been plated using incorrect antibiotic selection | If transformed E. coli cells from Step 21 were stored, replate cells on LB plates containing the appropriate antibiotics. If recovered cells were not stored, retransform Golden Gate reaction from Step 20 |
| Ligase activity may be compromised | Repeat Step 20 with fresh T4 DNA ligase and buffer | ||
| A component from the Golden Gate reaction mixture on Step 20 was omitted | Repeat Step 20, ensuring that each component is added to the reaction mixture | ||
| No visible growth of E. coli in liquid culture | Incorrect antibiotic was used in setting up the overnight culture | Create a new overnight culture using E. coli plates from Step 21, ensuring the correct antibiotic is used for selection | |
| Incorrect antibiotic was used when plating the recovered cells in Step 21 | Precursor backbones can be transformed and selected on solid LB medium if carbenicillin is used. If possible, replate stored cells from Step 21 on medium containing appropriate antibiotics, otherwise retransform Golden Gate reactions from Step 20 | ||
| T-DNA vector is missing a module used in the Golden Gate reaction | A component from the Golden Gate reaction mixture on Step 20 was omitted | Repeat Step 20, ensuring that each component is added to the reaction mixture | |
| A module used in the Golden Gate reaction on Step 20 has a mutation in the overhang | Determine the DNA sequence of overhangs on modules used in Step 20 | ||
| 26 | No visible growth of Agrobacterium colonies after 24 h at 28 °C | Incorrect antibiotic was used plating the recovered Agrobacterium cells in Step 24 | Refer to Critical step information for Step 24 for the correct antibiotic needed for each strain suggested in the reagents list |
Table 2 |.
Troubleshooting for Procedure 2: DD
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
|
| |||
| 17 | Plant death | Overly aggressive removal of tissue | When initially removing shoots, retain supporting leaves associated with each injection site. Each plant should retain two or three supporting leaves in total |
| 18 | Plant does not form any new shoots after initial removal of existing wild-type (WT) meristems | Some species may be recalcitrant to conversion of terminally differentiated somatic cells to meristematic stem cells | Intractable species may have physical or regulatory barriers that limit application of this protocol with the DRs described |
| De novo shoots experience tissue death, meristem abortion or sterility | Each plant species will have different sensitivity to overexpression of DRs | Consider using a different promoter to drive expression of DRs to obtain transgenic lines of interest | |
| High variability of shooting between induction groups or positional bias of shoot induction in growth chambers | Shooting in plants has been observed to be highly influenced by outside environmental factors such as light intensity, light spectrum, light duration, humidity, micro- and macronutrient availability, etc. All of the above can highly influence shoot organogenesis and morphogenesis | Efforts should be made to optimize growth conditions for shooting and harmonize conditions across experimental groups for reduced variability | |
| 21 | All shoots from injection sites are WT, and no transgenesis is observed | Apical dominance of residual WT shoots may inhibit de novo shoot formation | Consider lengthening the time period over which initial shoots are culled from the parent plant |
| The vast majority of shoots from injection sites are WT with limited transgenesis | Set culling period outlined in Step 18 may be too short for plant species | Consider lengthening the culling period to remove WT shoots | |
| No observable editing is found in tissues verified to be transgenic by reporter expression | Low expression of gene-editing reagents | Validate expression of gene-editing reagents in the plant species of interest by leaf infiltration | |
| Molecular screening of edits shows a distribution of edits that would not be likely for fixed mutations at the target site (even after considering the number of homologous alleles amplified by PCR screening) | Chimerism may be present that involves different fixed mutations across cell and tissue layers | Consider screening tissue at a later developmental stage or in the next generation | |
Table 3 |.
Troubleshooting for Procedure 3: Fast-TrACC
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
|
| |||
| 27 | Seedling/tissue death | Agrobacterium exposure is likely too high | Reduce starting bacterial concentration, co-culture time or both |
| 38 | No shoot formation from callus-like growths | Growths are slow to transition to shoots | Removing growths from the source plant and transferring them to solid 1/2 MS plates can promote shooting |
| 49 | No root regeneration from excised shoots | Calcification of the shoot base preventing root formation | Cut off the hardened base and transfer to new rooting medium |
| 54 | Plantlets dying after transfer | Plants were moved from root regeneration medium too early | Wait until more of a root system has been established. This will help the plantlet survive the stress of the transfer |
Timing
In addition, Figs. 5 and 6 illustrate the timelines for the DD and Fast-TrACC methods, respectively.
Fig. 5 |. Timeline for DD (Procedure 2).

Steps 1–5, N. benthamiana seeds are planted in soil and allowed to grow to maturity as indicated by formation of axillary shoots. Step 14, observable shoot meristems are removed from mature plants. T-DNA vectors are assembled in Procedure 1 that contain editing reagents, reporters or transgenes of interest. These T-DNA vectors are transformed into Agrobacterium to create −80 °C freezer stocks. Steps 6–13, Agrobacterium and liquid cultures are prepared for injections. Steps 15 and 16, the Agrobacterium cultures are injected into trimmed plants at sites where meristems were removed. Step 17, shoots that form in the first 20 d are removed. Step 18, de novo shoots form that are induced by DRs. Steps 19–21, shoots are screened for editing or transgenesis by analyzing DNA prepared from a leaf punch.
Fig. 6 |. Timeline for Fast-TrACC (Procedure 3).

Steps 1–8, sterile seeds are plated in 1/2 MS liquid medium. Step 9, the seeds are left to germinate until the cotyledons have emerged. T-DNA vectors are assembled in Procedure 1 that contain editing reagents, reporters or transgenes of interest. These T-DNA vectors are transformed into Agrobacterium to create −80 °C freezer stocks. Steps 10–18, Agrobacterium and liquid cultures are prepared for co-cultivation steps. Steps 19–27, Agrobacterium infection of seedlings in co-cultivation medium. Steps 28–33, after two d of co-culture, the seedlings are rinsed and moved to a new plate. Seedlings are left to grow for 2 weeks, during which time the ectopic expression of the DRs initiates de novo growths. Steps 34–37, newly emerging true leaves are trimmed, and new medium is added to the plates. Step 38, after roughly 20 d, de novo growths should be visible. Steps 39–46, once shoot-like structures form, they can be maintained on the seedling until they reach a size amenable for transfer to root-inducing medium. Alternatively, the shoot-like growths can be transferred to solid 1/2 MS medium until they reach a size amenable for transfer to root-inducing medium. Steps 47–49, shoot-like growths with leaflets are moved to root-inducing medium. Steps 50–54 plantlets with roots are transferred to soil. Steps 55–57, Shoots are screened for genetic modifications.
Procedure 1: modular Golden Gate assembly of T-DNA vector
Steps 1–3, design sgRNA(s) for target gene: 1 day
Steps 4–14, cloning synthesized oligos into tRNA MoClo compatible vectors: 3 d
Steps 15–18, construction of module B – guide: 3 d
Steps 19–21, assembly of T-DNA vector: 3 d
Steps 22–27, transformation into Agrobacterium: 3 d
Procedure 2: DD
Steps 1–5, plant selection and development: 4–8 weeks
Steps 6–13, Agrobacterium preparation for DD: 3 d
Steps 14–16, plant preparation and DD of Agrobacterium: ~2 h
Step 17, plant maintenance for DD: 20 d post inoculation
Step 18, de novo shoot formation for DD: 2–4 weeks
Steps 19–21, screening de novo shoot for genetic modifications in DD: variable
Procedure 3: Fast-TrACC
Steps 1–9, seed surface sterilization and germination for Fast-TrACC: 4–5 d
Steps 10–18, Agrobacterium preparation for Fast-TrACC: 4 d
Steps 19–27, Agrobacterium co-cultivation for Fast-TrACC: 2 d
Steps 28–33, transfer to growth development plates: 1 day (hands-on), 14 d (growth)
Steps 34–38, seedling maintenance: 1 day (hands-on), 6 d (growth)
Steps 39–43, shoot formation: 2 d to 2 weeks
Steps 44–46, growth cultures: every 2 weeks
Steps 47–49, root induction: 2–3 weeks
Steps 50–54, soil transfer of T0 plants: 1 day
Steps 55–57, screening de novo shoots for genetic modifications in Fast-TrACC: variable
Anticipated results
DD
Shooting will occur from preexisting primordial axillary meristems within 1–2d of trimming. These shoots should be continuously removed for the first 20 d. After 20 d, shoots arising from the delivery site will be a mixed population of spontaneously formed wild-type shoots and DR-induced shoots, which may harbor a genomic modification of interest. Transgenic shoots will begin to appear within 14–21 d of inoculation and will have a spectrum of phenotypes (Fig. 3f–h). Transgenic tissues that highly express DRs may experience tissue death, meristem abortion or sterility. Shoots induced by DRs without constitutive expression of the DR construct typically display no abnormal phenotypes. Transgenic shoots may be observed with as few as two or three plants, and the frequency depends on the number of injection sites per plant. Nontransgenic, edited meristems have been observed in N. benthamiana with as few as six plants (each plant harboring ~3 injection sites)6. Outcomes may vary due to growth conditions, plant age and delivery/expression of transgenes. As such, a single site to which ipt is delivered may produce from 0 to 20 individual shoots. Individual shoots from a given delivery site may be genetically unique with respect to transgene integration and targeting editing outcomes.
Fast-TrACC
Approximately 12 d after treatment with Agrobacterium, globular growths begin to appear, predominantly on the cotyledons of N. benthamiana seedlings. Over the next 10–14 d, the growths continue to progress, either in an undifferentiated callus-like state or they begin to form differentiated tissues, such as leaflets (Fig. 4e,f). By delivering both Wus2 and ipt, typically ~2–50% of the total growths progress to form shoots; if gene-editing reagents are included, ~25% of these shoots are edited6. Therefore, with a sgRNA targeting phytoene desaturase (PDS), inoculation of 24 N. benthamiana seedlings should yield roughly 20 de novo shoots, with ~5 of these exhibiting edits at PDS. For other genomic targets, the editing frequency will depend on the cutting efficiency of the sgRNA.
Data availability
The main data discussed in this protocol are available in the supporting primary research papers (https://doi.org/10.1038/s41587-019-0337-2) and (https://doi.org/10.3389/fgeed.2020.621710).
Supplementary Material
Acknowledgements
J.P.C. was supported by an NSF postdoctoral fellowship in biology (grant no. 2010445). M.F.M. was funded from NIGMS T32-GM008347.
Footnotes
Competing interests
M.F.M., R.A.N. and D.F.V. are named inventors on a patent application describing the DD and Fast-TrACC methods, which was filed by the University of Minnesota. D.F.V. consults for Calyxt, an agricultural biotechnology company that uses gene editing to create new crop varieties. All other authors have no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-022-00749-9.
Peer review information Nature Protocols thanks Dipankar Chakrabort, Peter Waterhouse and Yongliang Zhang for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Altpeter F et al. Advancing crop transformation in the era of genome editing. Plant Cell 28, 1510–1520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowe K et al. Rapid genotype ‘independent’ Zea mays L. (maize) transformation via direct somatic embryogenesis. Vitr. Cell. Dev. Biol. Plant 54, 240–252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson-Vasilchik K, Hague J, Mookkan M, Zhang ZJ & Kausch A Transformation of recalcitrant sorghum varieties facilitated by Baby Boom and Wuschel2. Curr. Protoc. Plant Biol. 3, e20076 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Gordon-Kamm B et al. Using morphogenic genes to improve recovery and regeneration of transgenic plants. Plants Basel Switz. 8, 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowe K et al. Morphogenic regulators Baby boom and Wuschel improve monocot transformation. Plant Cell 28, 1998–2015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maher MF et al. Plant gene editing through de novo induction of meristems. Nat. Biotechnol. 38, 84–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie K, Minkenberg B & Yang Y Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl Acad. Sci. USA 112, 3570–3575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Čermák T et al. A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29, 1196–1217 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber E, Engler C, Gruetzner R, Werner S & Marillonnet S A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 6, e16765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baltes NJ, Gil-Humanes J, Cermak T, Atkins PA & Voytas DF DNA replicons for plant genome engineering. Plant Cell 26, 151–163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon-Kamm B et al. Strategies for CRISPR/Cas9-mediated genome editing: from delivery to production of modified plants. Berleigh Dodds Ser. Agric. Sci. 1–36 (2021). [Google Scholar]
- 13.Ishida Y, Hiei Y & Komari T Agrobacterium-mediated transformation of maize. Nature 2, 1614–1621 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Parrot WA et al. Recovery of primary transformants of soybean. Plant Cell Rep. 7, 615–617 (1989). [DOI] [PubMed] [Google Scholar]
- 15.Yan B et al. Agrobacterium tumefaciens-mediated transformation of soybean [Glycine max (L.) Merril.] using immature zygotic cotyledon explants. Plant Cell Rep. 19, 1090–1097 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Matsuda N et al. Development of an Agrobaterium-mediated transformation method for pear (Pyrus communis L.) with leaf-section and axillary shoot-meristem explants. Plant Cell Rep. 24, 45–51 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Engler DE & Grogan RG Isolation, culture and regeneration of lettuce leaf mesophyll protoplasts. Plant Sci. Lett. 28, 223–229 (1983). [Google Scholar]
- 18.Li J et al. Multiplexed, targeted gene editing in Nicotiana benthamiana for glyco-engineering and monoclonalantibody production. Plant Biotechnol. J. 14, 533–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z et al. High frequency generation of fertile transgenic rice plants after PEG-mediated protoplast transformation. Plant Mol. Biol. Rep. 8, 276–291 (1990). [Google Scholar]
- 20.Rao KS et al. In planta transformation of pigeon pea: a method to overcome recalcitrancy of the crop to regeneration in vitro. Physiol. Mol. Biol. Plants 14, 321–328 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganguly S et al. Development of transgenic pigeonpea using high throughput plumular meristem transformation method. Plant Cell Tissue Org. Cult. 135, 73–83 (2018). [Google Scholar]
- 22.Ganguly S et al. Plumular meristem transformation system for chickpea: an efficient method to overcome recalcitrant tissue culture responses. Plant Cell Tissue Org. Cult. 142, 493–504 (2020). [Google Scholar]
- 23.Sparkes IA, Runions J, Kearns A & Hawes C Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1, 2019–2025 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Barton MK Twenty years on: the inner workings of the shoot apical meristem, a developmental dynamo. Dev. Biol. 341, 95–113 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Hoerster G et al. Use of non-integrating Zm-Wus2 vectors to enhance maize transformation: non-integrating WUS2 enhances transformation. Vitr. Cell. Dev. Biol. Plant 56, 265–279 (2020). [Google Scholar]
- 26.Doyle JJ & Doyle JL A rapid isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987). [Google Scholar]
- 27.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data discussed in this protocol are available in the supporting primary research papers (https://doi.org/10.1038/s41587-019-0337-2) and (https://doi.org/10.3389/fgeed.2020.621710).