

Abstract
Summary: Bacterial genomes are simpler than mammalian ones, and yet assembling the former from the data currently generated by high-throughput short-read sequencing machines still results in hundreds of contigs. To improve assembly quality, recent studies have utilized longer Pacific Biosciences (PacBio) reads or jumping libraries to connect contigs into larger scaffolds or help assemblers resolve ambiguities in repetitive regions of the genome. However, their popularity in contemporary genomic research is still limited by high cost and error rates. In this work, we explore the possibility of improving assemblies by using complete genomes from closely related species/strains. We present Ragout, a genome rearrangement approach, to address this problem. In contrast with most reference-guided algorithms, where only one reference genome is used, Ragout uses multiple references along with the evolutionary relationship among these references in order to determine the correct order of the contigs. Additionally, Ragout uses the assembly graph and multi-scale synteny blocks to reduce assembly gaps caused by small contigs from the input assembly. In simulations as well as real datasets, we believe that for common bacterial species, where many complete genome sequences from related strains have been available, the current high-throughput short-read sequencing paradigm is sufficient to obtain a single high-quality scaffold for each chromosome.
Availability: The Ragout software is freely available at: https://github.com/fenderglass/Ragout.
Contact: spham@salk.edu
1 INTRODUCTION
The recent proliferation of next-generation sequencing with short reads has enabled many new experimental opportunities, but it has also raised formidable computational challenges in genome assembly. Even for relatively simple bacterial genomes, their assemblies from current generation of high-throughput short reads are still fragmented with hundreds of contigs. To improve the assembly’s quality, recent studies have utilized longer Pacific Biosciences (PacBio) reads or jumping libraries to connect contigs into larger scaffolds or help assemblers resolve ambiguities in repetitive regions of the genome (Bashir, 2012; Deshpande, 2013; Koren, 2012). However, their popularity in current genomic research is still limited by high cost and error rates.
When a related genome is available, an alternative approach is to use this genome to guide the assembly of the target genome, in a method called ‘reference-assisted assembly’. The first reference-assisted assembly tools aligned contigs against the reference and ordered them according to their positions in the reference genome. While this approach is still commonly used, it introduces errors when structural variations between the reference and the assembled (target) genome are present. In an attempt to address this problem, Gaul and Blanchette (Gaul and Blanchette, 2006) formulated the contig ordering problem, which attempts to order contigs so that the 2-break distance (DCJ distance) (Alekseyev and Pevzner, 2009; Bergeron et al., 2006) between the resulting scaffold and the reference genome is minimized. This formulation has been further applied in some reference-guided assembly tools (Richter et al., 2007; Rissman et al., 2009). Unfortunately, while the approach is theoretically sound, these tools still generate erroneous scaffolds when there are rearrangements between the target and reference genomes. This poses an important question: is a single reference genome sufficient to obtain a single scaffold (for each chromosome) without errors?
Recently, Kim et al. (2013) introduced RACA software, which made an important step toward reliable reconstruction of the target (assembled) genome. In contrast to other tools, which use only one reference, RACA utilizes a reference as well as multiple outgroups to guide the assembly. This approach proved to be valuable, since the adjacency information in the outgroups can also help infer the adjacencies in the target assembly.
Although RACA marked an important advancement in the reference-guided assembly problem, it still suffers some limitations. First, RACA uses information from outgroup genomes, but it heavily relies on a single reference. As with any genome rearrangement tools, RACA decomposes these sequences into synteny blocks. However, rather than constructing synteny blocks by considering all input sequences, RACA constructs synteny blocks based on pairwise sequence alignment against only the reference genome. This approach, in some cases, cannot detect synteny blocks (Pham and Pevzner, 2010) and also raises the question of what to do with assembly sequences (contigs) that do not align against the reference. Second, unlike synteny blocks constructed from complete genomes, synteny blocks constructed in the presence of contigs can be fragmented, since assemblies usually have contigs of various lengths. Constructing synteny blocks from fragmented assemblies raises a problem: on which scale should synteny blocks be constructed? If one constructs large-scale synteny blocks, then small and fragmented synteny blocks (within small contigs) are not considered, thus leading to gaps in the assembly. On the other hand, if one constructs small-scale synteny blocks, then the rearrangement analysis becomes harder, since smaller synteny blocks are more likely to exhibit structural variations and are also more susceptible to be incorrectly identified (i.e. false synteny blocks). This dilemma must be addressed in order to obtain high-quality scaffolds.
In this work, we present Ragout (Reference-Assisted Genome Ordering UTility), a genome rearrangement approach for reference-assisted assembly that can produce high-quality scaffolds with a small number of misordered contigs and high genome coverage. Rather than working with a single reference, Ragout uses multiple complete genomes from closely related species/strains. In contrast with most existing tools, in which only a fixed scale of synteny blocks is used, our algorithm works iteratively with different scales of synteny blocks and also utilizes the assembly graph to improve scaffolds.
We demonstrate that with multiple references, Ragout significantly improves the assembly of the target genome compared to existing tools. Through simulations as well as real datasets, we believe that for common bacterial species, for which many complete genome sequences from related strains have been made available, the current high-throughput short-read-sequencing paradigm is sufficient to assemble into a single high-quality scaffold. The Ragout software is freely available at: https://github.com/fenderglass/Ragout.
2 METHODS
2.1 Algorithm overview
Ragout takes as input:
an assembly (a set of contigs);
a set of related genomes; and
a phylogenetic tree of all the genomes (including the target assembly).
It outputs high-quality scaffolds in the form of ordered lists of contigs.
Ragout first decomposes the input sequences into synteny blocks using Sibelia software (Minkin et al., 2013). After this stage, these sequences are represented in the alphabet of synteny blocks instead of as strings of nucleotides. While each reference sequence is transformed into a single sequence of synteny blocks (a list of signed numbers), the assembly corresponds to multiple sequences of synteny blocks because the target genome has been fragmented into multiple contigs.
Due to this fragmentation, some adjacency information is missing. Ragout uses a genome-rearrangement approach to infer these missing adjacencies. Initially, we filter all repetitive blocks as well as blocks that are not present in the target assembly, since these kinds of synteny blocks are hurdles in most current genome-rearrangement algorithms. From the remaining blocks, we build an incomplete multi-color breakpoint graph, in which vertices correspond to the ends of synteny blocks and edges represent adjacencies between them. Ragout further solves the Half-breakpoint State Parsimony Problem to transform this graph to the ‘normal’ multi-color breakpoint graph (Alekseyev and Pevzner, 2009) by recovering missing adjacencies in the target assembly. Next, contigs are assembled into scaffolds with respect to the inferred adjacencies. The above procedure is repeated multiple times with different synteny block scales, and the resulting scaffolds in these iterations are reconciled into a single set of scaffolds. Afterwards, a refinement step is performed. In this step, small and repetitive contigs are recovered and inserted back into the scaffolds by using the adjacency information from the assembly graph. The pseudocode of Ragout is described in Algorithm 1.
Algorithm 1 Ragout pseudocode.
procedure Ragout(references, target, phylogeny, blockSizes)
assemblies
for all blockSize in blockSizes do
synBlocks ← RunSibelia(references, target, blockSize)
bpGraph ← BuildBreakpointGraph(synBlocks)
weightedGraph ← EdgesScore(bpGraph, phylogeny)
adjacencies ← MinPerfMatching(weightedGraph)
scaffolds ← BuildScaffolds(target, adjacencies)
Add(scaffolds, assemblies)
end for
scaffolds ← MergeIterations(assemlies)
assemblyGraph ← BuildAssemblyGraph(target)
scaffolds ← RefineScaffolds(scaffolds, assemblyGraph)
OutputScaffolds(scaffolds)
end procedure
Since the synteny block-reconstruction algorithm used in Ragout has been described in (Minkin et al., 2013), we will not describe it in this article. Below, we delve into the details of Ragout algorithm, assuming that synteny blocks have already been constructed.
2.2 A genome-rearrangement approach for recovering missing adjacencies
Given reference sequences and assembly in the alphabet of synteny blocks, our task is to recover the missing adjacencies of synteny blocks in the target assembly. While breakpoint graphs best capture adjacency information, they have thus far only been defined for complete genomes. Below, we introduce incomplete multi-color breakpoint graphs for presenting synteny block adjacencies in the assembly and reference sequences.
2.2.1 Incomplete multi-color breakpoint graphs
Given an assembly A and k reference sequences P1, … , Pk in the alphabet of synteny blocks B, we define the incomplete multi-color breakpoint graph BG(A, P1, … , Pk) = (V, E), where For each synteny block, there are two vertices in the graph which correspond to the tail and head of the block. Edges are undirected and colored by k + 1 colors. An edge connects vertices that correspond to heads/tails of adjacent synteny blocks and is colored by the corresponding color of the genome/assembly. To simplify the notation, we use red, P1, … , Pk to refer to the colors of edges, where red edges represent the adjacencies of synteny blocks in the target assembly A, and Pi represents the adjacencies of synteny blocks in genome Pi (see Fig. 1A).
Fig. 1.

(a) A breakpoint graph of three reference genomes and one assembly. The three reference genomes (Ref1, Ref2 and Ref3) are presented as cyclic permutations of synteny blocks: Ref1(blue): + 1 +2 +3 +4 +5, Ref2(green): + 1 +3 +4 +5 and Ref3(orange): + 1 − 4 – 3 + 5, respectively. The target assembly (red) is presented as four separated permutations (corresponds to four contigs): Target Assembly: (b) A phylogenetic tree representing the states of the half-breakpoint 5h. Each leaf is labeled by the state of the half-breakpoint 5h in the corresponding reference/target genome. (According to this tree, the state of 5h in the target genome is 4t, although the correct state of 5h in the target genome is unknown.)
Before constructing the graph, we filter synteny blocks from reference genomes that are not present in the target assembly, since these synteny blocks do not help to infer the adjacencies in the target genome. Also, we filter synteny blocks that have multiple copies within any sequences, since duplicated synteny blocks make further analysis in the breakpoint graph complicated.
After this filtering, the constructed graph has two important properties: (i) each vertex has at most one edge of each color (since all repetitive synteny blocks are removed), and (ii) each vertex corresponds to a certain synteny block in the target genome. Therefore, if the target genome were available, the set of all red edges would define a perfect matching in the graph. However, since the genome is fragmented into contigs, the adjacency information of the target genome at the vertices that correspond to the end of contigs is missing. The main task is to infer these missing red edges in the graph using other adjacencies from the reference genomes as well as using their evolutionary relationship in the form of a phylogenetic tree.
2.2.2 The phylogenetic tree
Let T be a rooted phylogenetic tree of the genomes A, P1, … , Pk. T consists of k + 1 leaves, (k – 1) internal nodes of degree three, and one node of degree two (called the root). Edges connecting two nodes are called branches. Branch length represents evolution time (evolution time is usually inferred from the number of nucleotide substitutions in sequence alignment).
A parsimonious model for rearrangements
Given a tree T with all of its k + 1 leaves represented as complete genomes (in the alphabet of synteny blocks), the parsimony procedure (Fitch, 1971; Sankoff, 1975) explains the descent of these various sequences from a common ancestor with the fewest number of changing operations, associated with a minimum cost value. When one of these genomes is divided into contigs, a possible formulation for recovering the target genome corresponds to finding a permutation of these contigs such that the parsimony procedure returns the lowest cost among all possible orderings of these contigs. Note that this is a double optimization. One step finds the most parsimonious explanation for a given configuration of the target genome, while the other step finds the configuration of the target genome having lowest cost. The usual (and more correct) choice for the allowed operation in the first optimization is the 2-break operation (also called a DCJ operation) since it can well approximate many real rearrangement operations (Alekseyev and Pevzner, 2009). However, accounting for such an operation will lead to an NP-complete problem (Ma et al., 2006). In this work, rather than using the 2-break operation, we study the parsimonious approach on individual breakpoints. While considering breakpoints individually may not adequately reflect the reality of rearrangements (since each rearrangement uses at least two breakpoints), this method is computationally feasible and has been proven to be valuable in the problem of ancestral genome reconstruction (Ma et al., 2006).
In the breakpoint graph BG(A, Pi, … , Pk), we call each vertex a half-breakpoint (since a breakpoint involves two synteny block ends). For a given genome Pi, the state of a half-breakpoint vi is defined as the half-breakpoint adjacent to it, i.e. the vertex vj, such that the color of the edge (vi, vj) is Pi (color) in BG. Because of the first graph property described above, each half-breakpoint has at most one such state. If the synteny block corresponding to vi is missing in a genome, the state vi is void (see Fig. 1B).
Rather than using the parsimony procedure with 2-break operations, we study the parsimony procedure with respect to the change of half-breakpoint’s states for each vertex in the breakpoint graph. The cost associated with a state change along a branch b of length τ in the phylogenetic tree is Intuitively, the longer a branch, the more likely a half-breakpoint state can change along it, and therefore the smaller the corresponding cost. Next, we formulate the Half-breakpoint State Parsimony Problem in order to infer the evolutionary scenario of a particular half-breakpoint.
2.2.3 Half-breakpoint state parsimony
Given a half-breakpoint u and the phylogenetic tree T, each leaf of which is labeled by state(u) in each corresponding genome. Label the internal nodes of T in order to minimize the total cost needed to derive the leaves’ states from their common ancestor.
Below is a linear time algorithm for solving this problem. The solution for this problem mimics Sankoff’s dynamic programming algorithm for the weighted small parsimony problem. The optimality proof is perfectly analogous to Sankoff’s proof (Sankoff, 1975).
Input: for each leaf node of tree.
Output: for each internal node, along with the corresponding cost.
Objective function: st(v) = minimum score of the subtree rooted at vertex v if v has state t.
Initialization: for each leaf node l: st(l) = 0 if the state of leaf node is t, otherwise st(l) = ∞.
- Recursion: The score at each vertex is based on the scores of its children:

Termination:
Complexity: O(n·d), where n is the number of leaves and d is the number of possible states.
When all the internal nodes of the tree have already been labeled, the cost of half-breakpoint state parsimony of the half-breakpoint u can be calculated by summing the cost in all the branches that the state changes.
| (1) |
2.2.4 Recovering missing adjacencies
Since the half-breakpoint state parsimony problem can be solved efficiently, the remaining question is to recover all missing adjacencies such that —the total cost of all half-breakpoints in G—is minimal. Since we filtered all duplicated synteny blocks as well as synteny blocks that do not appear in the target assembly, adjacencies in the target genome define a perfect matching in the graph. The cost of this matching is defined to be the sum of the half-breakpoint parsimony cost of all vertices (half-breakpoints).
For each vertex that is not incident to a red edge in the breakpoint graph, the Ragout algorithm finds the cost for the half-breakpoint state parsimony problem on each of its possible states. These states are chosen from the vertex’s adjacent vertices in the breakpoint graph. Since choosing a state for a particular half-breakpoint corresponds to choosing an edge incident to it, for each edge in the breakpoint graph, two such cost values are calculated. We assign the weight of each edge as the sum of these two values. As some adjacencies in the target genome are already known (vertices incident to a red edge), we can also remove those vertices from the graph before applying the Blossom algorithm to find the perfect matching with minimum cost (in O(|V |4) time) and add corresponding edges into the final matching afterwards. This matching defines the optimal adjacencies in the target genome.
2.2.5 Building scaffolds
Finally, we order contigs into scaffolds. Starting from one contig, we try to extend it in both the forward and backward directions, using the inferred adjacencies from the matching above.
2.3 The choice of minimum synteny block size and iterative assembly
Since synteny block reconstruction is a parameter-dependent procedure, and the choice of parameters depends on the ‘synteny block scale’ required in each particular application, an important question to ask is: which scale of synteny blocks should one use for reference-assisted assembly? We argue that using a single scale of synteny blocks is not sufficient to obtain high-quality scaffolds. If one constructs large-scale synteny blocks, then small and fragmented synteny blocks (coming from small contigs and micro-rearrangements) are not considered, thus leading to gaps in the assembly. On the other hand, if one constructs small-scale synteny blocks, then rearrangement analysis becomes more difficult, since small-scale blocks are more likely to exhibit structural variations and are also more susceptible to be incorrectly identified (false synteny blocks). To resolve this dilemma, we use multiple synteny block scales in order to build multiple scaffolds and then reconcile these scaffolds.
Consider two scaffolds As and Aw constructed from large- and small-scales of synteny blocks, respectively. A contig is called strong if it is in the scaffold As and called weak if it is in Aw and not in As. Two scaffolds As and Aw are called consistent if every pair of adjacent contigs in As is either adjacent or separated by only weak contigs in Aw. Although the order of contigs in As is more reliable than in Aw, there are many small synteny blocks that do not reveal in As and only appear in the ‘finer’ scaffold Aw. We therefore rely on the ‘skeleton of contigs’ in As and insert smaller contigs from Aw if they are consistent (see Fig. 2).
Fig. 2.

Merging two scaffolds As and Aw built from two different synteny scales into a scaffold M. Yellow rectangles represent weak contigs. (a) As and Aw are consistent. (b) As and Aw are not consistent
The merged scaffold is therefore consistent with As. We successively apply the described procedure to scaffolds in different stages (sorted by the scale of synteny blocks) until we reach the stage with the smallest scale.
2.4 Refinement with the assembly graph
While the iterative procedure attempts to maximize the number of contigs used to build the scaffold, there are still some certain types of contigs that cannot be utilized in that stage. These contigs include: (i) contigs that are shorter than minimum synteny block size that existing synteny block tools can detect (several hundred nucleotides), (ii) contigs contained only in the target genome and (iii) repetitive contigs. Such fragments are not considered in the rearrangement analysis and will therefore not appear in the merged scaffolds. To add these fragments, we need to use the assembly graph, which has been output by short-read assemblers and can also be independently constructed from input contigs. The genome traverses the graph with a certain unknown path. However, since initial scaffolds are now available, we can use these scaffolds to restore small or repetitive fragments. This problem is analogous to the problem of repeat resolution in short-read assembly. Instead of paired-read information, we have pairs of adjacent contigs built during the rearrangement analysis stage.
Given an assembly graph and a set of merged scaffolds from the previous step of the algorithm, for each pair of consecutive contigs from these scaffolds we find all possible paths connecting them in the assembly graph that do not contain contigs from the scaffold. If there exists only one such path, we insert all the intermediate contigs along this path between the two contigs (see Fig. 3). This procedure significantly improves the number of used contigs in most datasets.
Fig. 3.
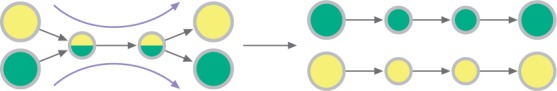
Refinement with the assembly graph. The procedure fills scaffold gaps with small contigs. The big circles illustrate contigs with known order, while small ones correspond to contigs that were not considered in rearrangement analysis
3 RESULTS
We benchmarked Ragout against other reference-assisted assembly tools [Mauve Contig Mover (Rissman et al., 2009), OSLay (Richter et al., 2007), RACA (Kim et al., 2013)] on one simulated and three real bacterial datasets. Since the complete sequences of target genomes are available, we can also assess the quality of the resulting scaffolds by the number of misordered contigs, scaffold gaps and coverage.
As each output scaffold is an ordered list of contigs, we define the number of misordered contigs as the minimum number of contigs in the scaffolds whose mapping positions or directions are not consistent with the mapping positions and directions of the contigs before and after them. Also, we define the number of gaps in a scaffold as the number of contig pairs that are adjacent in a scaffold, but are separated by some other contigs when we map all contigs to the reference genome. The coverage is defined as the total number of aligned bases against the reference, divided by the genome size.
Mauve Contig Mover and OSLay were run with parameters recommended for bacterial genomes. For Ragout, we ran three iterations with the corresponding minimum synteny block sizes: 5000, 500, 100, as they are the default scales used in Sibelia (Minkin et al., 2013) for bacterial genomes. Since RACA works with only one synteny block size, we chose the maximal synteny block scale (most reliable).
3.1 Assembly using one closely related reference
First, we benchmark these tools on an easy case in which the target and reference genomes do not exhibit any structural variations. In this situation, one reference is sufficient to obtain the correct assembly. This also allows us to compare Ragout with MCM and OSLay, which can work only with one reference.
The dataset consists of two different Escherichia coli strains: DH1 (NC_017625) as the reference and K-12 subs. MG1655 (NC_000913) as the target. The contigs were assembled with SPAdes assembler (Bankevich et al., 2012). The results can be seen in Table 1. Ragout and MCM are able to recover one complete scaffold, while OSLay outputs eight scaffolds. The quality of Ragout’s assembly without refinement is quite similar to MCM, but with refinement Ragout uses significantly more contigs and produces fewer gaps in the final scaffolds.
Table 1.
Comparison of different tools using one reference
| Ragout | MCM | OSLay | |
|---|---|---|---|
| Scaffolds | 1 (1) | 1 | 8 |
| Coverage | 97.9 (97.6) | 97.6 | 96.7 |
| Ordered contigs | 129 (79) | 77 | 80 |
| Gaps | 52 (71) | 73 | 61 |
| Misordered | 0 (0) | 0 | 1 |
All tools were run with their default parameters. For Ragout, results are given both with and without (in brackets) refinement. The total number of the contigs is 156. Initial assembly coverage is 98.18%.
3.2 Assembly of one chromosome using multiple references
Next, we want to address a more problematic case in which multiple references are available, but each of these references exhibits structural variations comparing to the target genome. The dataset contains five different Helicobacter Pylori strains: Puno120 (NC_017378), ELS37 (NC_017063), Gambia94/24 (NC_017371) and G27 (NC_011333) as references and SJM180 (NC_014560) as the target. The corresponding phylogenetic tree is shown on Figure 4A. The dot plots showing the rearrangements can be seen in Figure 5. Contigs were assembled using ABYSS (Simpson et al., 2009).
Fig. 4.

Phylogenetic trees. (a) Heclicobacter Pylori with SJM180 as target. (b) Vibrio Cholerae with H1 as target. (c) Staphylococcus Aureus with USA300 as target. (d) Simulated genomes. Solid branches contain all types of rearrangements, while dashed branches contain only indels
Fig. 5.

(a−d) Dot plots of H.Pylori references versus target genomes. (e) Dot plot of Ragout’s scaffold versus the target genome showing a perfect diagonal line for visual verification
First, we run Ragout as well as OSLay and MCM on each of the available references separately to illustrate that the ‘usual’ assisted assembly with one reference is insufficient for the current case. Every tool produces a certain amount of misordered contigs, which can be explained by the structural divergence between the reference and the target (see Table 2).
Table 2.
Comparison with MCM and OSLay using one H. Pylori reference showing misordered contigs
| Reference | Scaffolds | Ordered | Misordered | Coverage |
|---|---|---|---|---|
| Mauve contig mover | ||||
| G27 | 1 | 53 | 7 | 97.9 |
| Gambia94/24 | 1 | 54 | 8 | 97.9 |
| ELS37 | 1 | 45 | 9 | 98.0 |
| Puno120 | 1 | 56 | 11 | 98.0 |
| OSLay | ||||
| G27 | 5 | 50 | 1 | 96.2 |
| Gambia94/24 | 8 | 49 | 3 | 96.4 |
| ELS37 | 6 | 53 | 3 | 98.0 |
| Puno120 | 8 | 51 | 2 | 87.0 |
| Ragout | ||||
| G27 | 1 (1) | 91 (50) | 4 (4) | 97.7 (97.4) |
| Gambia94/24 | 2 (2) | 83 (45) | 7 (7) | 96.8 (96.6) |
| ELS37 | 1 (1) | 102 (56) | 4 (3) | 98.1 (97.5) |
| Puno120 | 2 (2) | 92 (49) | 6 (6) | 97.2 (96.9) |
All tools were run with their default parameters. For Ragout, results are given both with and without (in brackets) refinement. The total number of contigs is 183. Initial assembly coverage is 98.57%.
Since OSLay and MCM can only run with one reference, we use Ragout and RACA to benchmark different sets of multiple references (see Table 3). For RACA, G27 was chosen to be the reference and others were treated as outgroups. Both tools have misordered contigs when using three or fewer references. Ragout is able to infer the correct order of the contigs with the set of four references, while RACA still has some misordered contigs. Also, Ragout outputs the assembly with better coverage and fewer gaps.
Table 3.
Comparison of Ragout and RACA on H.pylori using multiple references
| References | Scaffolds | Coverage | Ordered | Gaps | Misordered |
|---|---|---|---|---|---|
| Ragout | |||||
| G27, ELS37 | 2 (2) | 97.8 (97.6) | 95 (53) | 22 (39) | 1 (1) |
| G27, Puno120, ELS37 | 1 (1) | 97.8 (97.6) | 95 (53) | 21 (36) | 1 (1) |
| G27, Puno120, Gambia94/24, ELS37 | 1 (1) | 97.6 (97.3) | 93 (46) | 22 (38) | 0 (0) |
| RACA | |||||
| G27, ELS37 | 3 | 83.6 | 35 | 29 | 2 |
| G27, Puno120, ELS37 | 2 | 83.6 | 35 | 30 | 1 |
| G27, Puno120, Gambia94/24, ELS37 | 2 | 83.8 | 35 | 31 | 1 |
All tools were run with their default parameters. For Ragout, results are given both with and without (in brackets) refinement. The total number of contigs is 183. Initial assembly coverage is 98.57%.
3.3 Multiple chromosome assembly using multiple references
The next dataset was taken from a recent study (Bashir et al., 2012) in which long PacBio reads were used to obtain the complete de novo assembly of Vibrio Cholerae H1 str. (AKGH01000001). Here we want to show that with multiple related references (even though these references and the target genome have structural variations), complete scaffolds with high quality can also be obtained from only short-read data. We used SPAdes to assemble non-paired Illumina reads with read length 40 bp. Three references were chosen so that each of them would have rearrangements in at least one chromosome compared to the target genome: O1 Biovar (AE003852), O1 Inaba (CM001785) and O395 (CP001235). See Figure 6 for the dot-plots. The phylogenetic tree is shown on Figure 4(B).
Fig. 6.

Dot plots of different chromosomes of V.Cholerae references (a–c) versus the corresponding chromosomes of the target genome showing rearrangements
Using three references, Ragout was able to correctly reconstruct two scaffolds that correspond to two V.Cholerae chromosomes (see Table 4). Refinement with the assembly graph significantly increases the number of utilized contigs.
Table 4.
Comparison of Ragout and RACA on V.cholerae using multiple references
| References | Scaffolds | Coverage | Ordered | Gaps | Misordered |
|---|---|---|---|---|---|
| Ragout | |||||
| O1 Inaba | 3 (3) | 95.3 (94.8) | 317 (185) | 41 (64) | 5 (3) |
| O1 Inaba, O1 biovar | 2 (2) | 95.5 (94.7) | 305 (179) | 46 (68) | 6 (4) |
| O1 Inaba, O1 biovar, O395 | 2 (2) | 95.5 (94.7) | 300 (174) | 46 (66) | 2 (0) |
| RACA | |||||
| O1 Inaba, O1 biovar | 6 | 85.8 | 124 | 51 | 0 |
| O1 Inaba, O1 biovar, O395 | 3 | 90.0 | 127 | 56 | 0 |
All tools were run with their default parameters. For Ragout, results are given both with and without (in brackets) refinement. The total number of contigs is 1407. Initial assembly coverage is 96.89%.
For RACA, O1 Inaba was chosen to be the reference and others were treated as outgroups. RACA outputs three correct scaffolds with three references and six scaffolds with two references. Ragout outperforms RACA by the quality of the assembly, both with and without refinement.
3.4 Assembly using structurally divergent references
Finally, we want to address the case when the references have a large number of structural variations. This case requires us to perform simulations because bacterial genomes usually have few rearrangements.
The phylogenetic tree that was used for simulations is shown on Figure 4(D). For the sake of simplicity, the tree is chosen to be symmetrical and can be treated both as an unrooted tree or as a rooted one with the target branch of infinitesimal length. On each of the four outer branches (incident to references), five reversals and five translocations were simulated. Additionally, to make the dataset more complex, we have simulated 10 indels on each of the tree branches, including all inner ones. Genomes were then decomposed into synteny blocks, and then the target genome was split into contigs, where each contig represents exactly one synteny block.
Our analysis includes three cases corresponding to when four, three or two of the simulated references are available. For each case, we have generated 100 different datasets. Since the phylogenetic tree is symmetric, the result does not depend on which particular reference is absent. We took E.coli K-12 str. MG1655 substr. (NC_000913) as a target and the number of synteny blocks in decomposition was 112 in average.
Next, we run Ragout on every dataset. The results of simulations can be seen in Figure 7, which clearly shows that the number of misordered contigs increases when some of the references are missing from the analysis. The errors in the case when all references are available can be explained by breakpoint reuse, which makes it impossible for the algorithm to distinguish overlapping rearrangements.
Fig. 7.

Correspondence of the number of available references with the number of misordered contigs for Ragout
To compare Ragout and RACA, we chose two datasets with four references, since each run of RACA requires a large number of manual preparations. Results can be seen in Table 5. In both cases, Ragout produces better assemblies than RACA; one possible explanation for this phenomenon is that RACA heavily relies on one particular reference.
Table 5.
Comparison of Ragout and RACA on simulated datasets
| Dataset 1 |
Dataset 2 |
|||
|---|---|---|---|---|
| Ragout | RACA | Ragout | RACA | |
| Scaffolds | 1 | 6 | 1 | 8 |
| Coverage | 95.3 | 83.5 | 94.5 | 75.4 |
| Ordered | 112 | 101 | 112 | 90 |
| Gaps | 3 | 6 | 11 | 5 |
| Misordered | 0 | 3 | 4 | 2 |
Ragout and RACA were run with synteny block size equal to 500 (the size is known from the simulations) without refinement. The total number of contigs is 114 in both cases. Initial assembly coverage is 100%.
3.5 Parameter choice and iterative assembly
We would like to benchmark Ragout with different parameters of iterative/non-iterative assembly. The dataset consists of five strains of Staphylococcus Aureus as references: COL (NC_002951), JKD6008 (NC_017341), N315 (NC_002745), RF112 (NC_007622) and USA300 (NC_007793) as the target. The phylogenetic tree is shown in Figure 4(C). Contigs were assembled from single-cell sequencing data using SPAdes.
The results of benchmarking can be seen in Table 6. As expected, the smaller size of a synteny block allows the algorithm to arrange more contigs, but analysis becomes more complicated. As a result, the algorithm produces some incorrect adjacencies which leads to misordered contigs and more fragmented assembly (in number of scaffolds). Next, iterative assembly was performed in three stages (5000, 500, 100). This assembly kept the complete scaffold from the first stage, while adding some smaller contigs from the second and third stages. Even though some misordered contigs could be carried to the merged scaffolds, these errors are local and do not violate the correct ‘skeleton’ of scaffolds.
Table 6.
Iterative assembly of S.Aureus
| SB size | 100 | 500 | 5000 | Iterative |
|---|---|---|---|---|
| Scaffolds | 5 | 2 | 1 | 1 |
| Coverage | 96.7 | 96.3 | 92.0 | 96.7 |
| Ordered | 108 | 91 | 62 | 89 |
| Gaps | 75 | 75 | 56 | 73 |
| Misordered | 1 | 0 | 0 | 1 |
Ragout was run with four S.Aureus references with different minimum synteny block sizes. Iterative assembly was performed with all previous sizes combined (5000, 500, 100). The total number of contigs is 767. Initial assembly coverage is 98.4%. We did not perform refinement with the assembly graph in order to focus on the effect of synteny block size.
4 DISCUSSION
In this article, we have presented Ragout, a package for improving assemblies using multiple complete references. We demonstrated that with multiple related genomes available, one can obtain a complete and high-quality scaffold for each chromosome using only high-throughput short-read sequencing. This marks an important improvement in genome assembly of short reads and even raises a question whether long PacBio reads or long jumping libraries are needed for genomic studies of common bacteria where multiple related references have been available.
The current version of Ragout uses Sibelia for synteny block reconstruction and therefore limits itself to bacterial genomes. When synteny blocks have been available, Ragout is fast and memory-efficient. We plan to make Ragout compatible with other synteny block generation tools that can work with mammalian genomes [e.g. Cactus aligner Paten et al. (2011)] and further extend Ragout to work with mammalian datasets. Another limitation of Ragout is that it only uses the assembly graph for recovering repetitive blocks or small contigs that could not be captured in synteny analysis. Therefore, it can make mistakes when rearrangements happened on the target branch. Since de Bruijn graphs can be transformed into breakpoint graphs and vice-versa, the de Bruijn graphs output from short-read assemblers can also be used for rearrangement analysis and we will focus on this issue in further studies.
ACKNOWLEDGEMENTS
We would like to thank Jaebum Kim for assisting us with the benchmark of RACA software. We are indebted to Phillip Compeau, Apua Paquola, Nitin Udpa, Shay Zakov, Nikolay Vyahhi and Han Do for revising the manuscript and for many helpful suggestions that significantly improve the paper.
Funding: NIH (grant number 1U41HG007234-01) and VP Foundation (grant number BI-2013-02), in part.
Conflict of Interest: none declared.
REFERENCES
- Alekseyev MA, Pevzner PA. Breakpoint graphs and ancestral genome reconstructions. Genome Res. 2009;19:943–957. doi: 10.1101/gr.082784.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A, et al. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012;30:701–707. doi: 10.1038/nbt.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir K, et al. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012;30:701–709. doi: 10.1038/nbt.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron A, et al. Proceedings of Algorithms in Bioinformatics. Springer; 2006. A unifying view of genome rearrangements; pp. 163–173. [Google Scholar]
- Deshpande V, et al. Proceedings of Algorithms in Bioinformatics. Springer; 2013. Cerulean: A hybrid assembly using high throughput short and long reads; pp. 349–363. [Google Scholar]
- Fitch WM. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Biol. 1971;20:406–416. [Google Scholar]
- Gaul E, Blanchette M. Proceedings of the Comparative Genomics. Springer; 2006. Ordering partially assembled genomes using gene arrangements; pp. 113–128. [Google Scholar]
- Kim J, et al. Reference-assisted chromosome assembly. Proc. Natl Acad. Sci. USA. 2013;110:1785–1790. doi: 10.1073/pnas.1220349110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, et al. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006;16:1557–1565. doi: 10.1101/gr.5383506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkin I, et al. Proceedings of Algorithms in Bioinformatics. Springer; 2013. Sibelia: A scalable and comprehensive synteny block generation tool for closely related microbial genomes; pp. 215–229. [Google Scholar]
- Paten B, et al. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011;21:1512–1528. doi: 10.1101/gr.123356.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham SK, Pevzner PA. Drimm-synteny: decomposing genomes into evolutionary conserved segments. Bioinformatics. 2010;26:2509–2516. doi: 10.1093/bioinformatics/btq465. [DOI] [PubMed] [Google Scholar]
- Richter DC, et al. Oslay: optimal syntenic layout of unfinished assemblies. Bioinformatics. 2007;23:1573–1579. doi: 10.1093/bioinformatics/btm153. [DOI] [PubMed] [Google Scholar]
- Rissman AI, et al. Reordering contigs of draft genomes using the mauve aligner. Bioinformatics. 2009;25:2071–2073. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 1975;28:35–42. [Google Scholar]
- Simpson JT, et al. Abyss: A parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]