关注
关注






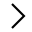
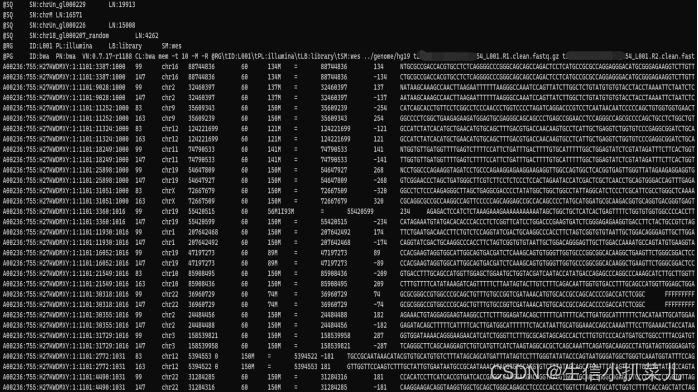
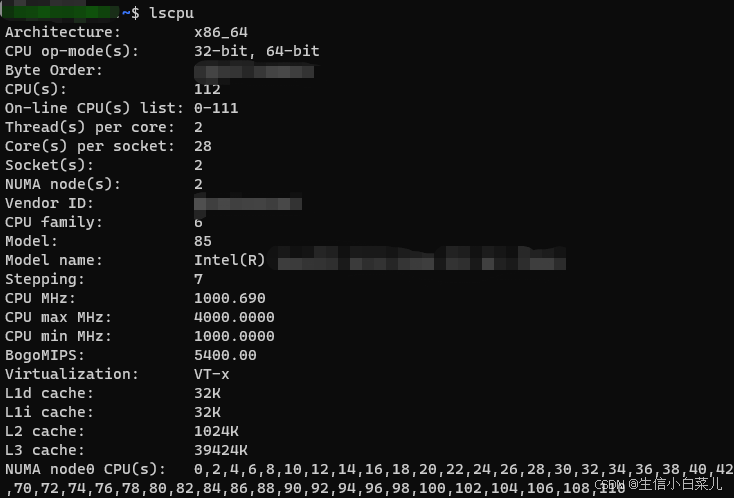
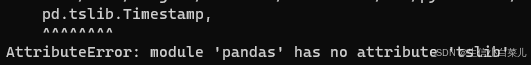
一文读懂全外显子组测序
下面是本人自学全外过程中总结的所有知识点。
TA关注的专栏 0
TA关注的收藏夹 0
TA关注的社区 2
TA参与的活动 0


AI 镜像开发实战征文活动
随着人工智能技术的飞速发展,AI 镜像开发逐渐成为技术领域的热点之一。Stable Diffusion 3.5 FP8 作为强大的文生图模型,为开发者提供了更高效的图像生成解决方案。为了推动 AI 镜像开发技术的交流与创新,我们特此发起本次征文活动,诚邀广大开发者分享在 Stable Diffusion 3.5 FP8 文生图方向的实战经验和创新应用 本次征文活动鼓励开发者围绕 Stable Diffusion 3.5 FP8 文生图方向,分享以下方面的内容: 1. 技术实践与优化 - Stable Diffusion 3.5 FP8 模型架构解析与优化技巧 - 文生图生成效果的提升方法与技巧 - 模型部署与加速策略,例如使用 Hugging Face、Diffusers 等工具 - 针对特定场景(例如二次元、写实风)的模型微调与定制化开发 2. 应用场景探索 - Stable Diffusion 3.5 FP8 在不同领域的应用案例分享,例如游戏设计、广告创意、艺术创作等 - 利用 Stable Diffusion 3.5 FP8 实现图像编辑、图像修复、图像增强等功能的探索 - 结合其他 AI 技术(例如 NLP、语音识别)构建更强大的应用 3. 创新应用与思考 - 基于 Stable Diffusion 3.5 FP8 的创新应用场景设计 - AI 镜像开发的未来发展方向的思考与展望 - 对 AI 镜像开发伦理、安全等问题的探讨