~ Stem-cell ‘tourists’ travel to where they have access to controversial stem-
cell therapies/ treatments.
~ Every baby born a decade from now will have its genetic code mapped at
birth predicts head of genomics company. Just because we can, does that
mean we should?
~ More docs tell pharma reps to keep out. Does this mean no more free post-
its?
~ Comprehensive reform bill that would have banned pharma gifts to docs voted
down in Colorado.
~ Experts say consumers should have more facts in drug ads so they can make informed decisions.
~ Glaxo to cut prices on drugs sold in poor countries. They will also invest
profits in building clinics in those countries.
~ The U.S. drug industry has shifted most of its clinical trials to overseas
sites raises serious ethical concerns.
~ U.S. court: No link between vaccines and autism.
~ Pfizer owes damages for bilking Wisconsin Medicaid.
~ EU governments have no right to conceal the location of field trials of genetically modified (GM) crops.
~ Men may be their own worst enemy when it comes to their health.
~ Women on the other hand …: Coffee drinking lowers women’s stroke risk. Ooooh, imagine a Starbucks ‘pharmacy’ on every corner …
~ BUT, too much soda can kill a girl’s kidneys.
~ Llama’s have unique antibodies that one day might be used to treat immune system diseases in humans.
~ Scientists have unraveled the genetic code of the common cold. Spectacular!
~ Decoy molecules drive cancer cells to suicide.
~Altered virus effectively delivers new gene to replace faulty one that causes CF and completely rids the lung of disease. I wonder if these researchers have seen I Am Legend?
~ Researchers have discovered that the good bacteria found in dairy products might also be an effective vehicle for an oral vaccine that can provide immunity to anthrax exposure.
~ A new study indicates that a pneumonia vaccine can significantly cut the risk of heart disease.
~ Oh, Baby: A prenatal link to Alzheimer's?
~ Doctors have identified two genetic mutations that control the growth and
development of malignant gliomas; maybe good news for brain tumor patients.
~ Cotton candy as a substrate to re-grow vascular tissue.
~ Biotechnology's potential barely exploited.
~ Stimulus package includes funds for comparison of the effectiveness of
medical treatments.
~ President Obama to lift ban on embryonic stem cell research soon.
~ Scientists and doctors try to qualm public fears about vaccines and autism.
~ Scientists preparing to storm Capitol Hill on March 25 (a.k.a. the million
scientist march?). Registration ends Feb. 23.
~ No European stem cell patent for spinal cord repair.
~ Retired nurse invents cough, sneeze cover. Maybe she can convince the
airlines to make these standard issue …
~ FDA approves new and improved treatment for gout (the first in 40 ~years!).
~ But agency second guessing another …Savient gout drug faces approval delay.
~ FDA orders Bayer to correct earlier claims in Yaz birth control ad.
~ FDA deliberately backed off of "Good Laboratory Practice" requirements for
medical device makers.
~FDA wants one strain changed for next flu vaccine.
~ Orphaned baby chimpanzees cared for by humans in a loving, attentive manner have been found to be more cognitively advanced than some human infants. But, then, is this really that weird? They do share over 99% of our DNA.
~ Parody: FDA Approves Depressant Drug For The Annoyingly Cheerful.
[Thank you to Lisa von Biela, JD candidate, 2009, UMN, Editor of the BioBlurb, from which this content is partially taken and edited. BioBlurb is a weekly electronic publication of the American Bar Association's Committee on Biotechnology, Section of Science & Technology Law. Archived issues of the BioBlurb, as well as further information about the Committee on Biotechnology, are available here.]
 As for my other objections, they are relatively simple from a technological standpoint. Unless technology has rapidly changed in the last few years, one of the major drawbacks of cochlear implants is that they do destroy any residual hearing. This is why many doctors suggest only implanting one ear, in case a better technological or biological solution comes along later down the line. So you are wedded to the device implants, and that technological level, for the remainder of your life.
As for my other objections, they are relatively simple from a technological standpoint. Unless technology has rapidly changed in the last few years, one of the major drawbacks of cochlear implants is that they do destroy any residual hearing. This is why many doctors suggest only implanting one ear, in case a better technological or biological solution comes along later down the line. So you are wedded to the device implants, and that technological level, for the remainder of your life.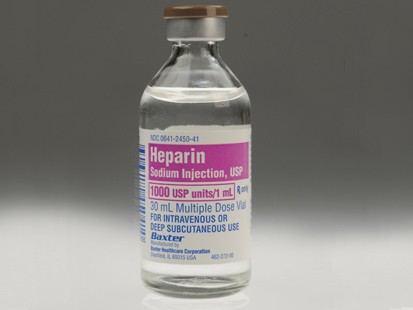 For the sake of people susceptible to earworms everywhere, I won't actually parody the Soft Cell song further than using it as a title here. And as alliterative and 80s-referential as the title is, it's also accurate: in recent weeks, almost two dozen people have died, and their deaths
For the sake of people susceptible to earworms everywhere, I won't actually parody the Soft Cell song further than using it as a title here. And as alliterative and 80s-referential as the title is, it's also accurate: in recent weeks, almost two dozen people have died, and their deaths  It would seem that the newest thing in anti-aging is nanosomes. Nanospheres.
It would seem that the newest thing in anti-aging is nanosomes. Nanospheres. 
 A state-owned Chinese pharmaceutical company is at the heart of an international drug scandal after it's been revealed that over 200 patients were paralyzed or otherwise hurt by tainted leukemia drugs last summer. But in what we sometimes cynical Westerner's might
A state-owned Chinese pharmaceutical company is at the heart of an international drug scandal after it's been revealed that over 200 patients were paralyzed or otherwise hurt by tainted leukemia drugs last summer. But in what we sometimes cynical Westerner's might  According to
According to 







